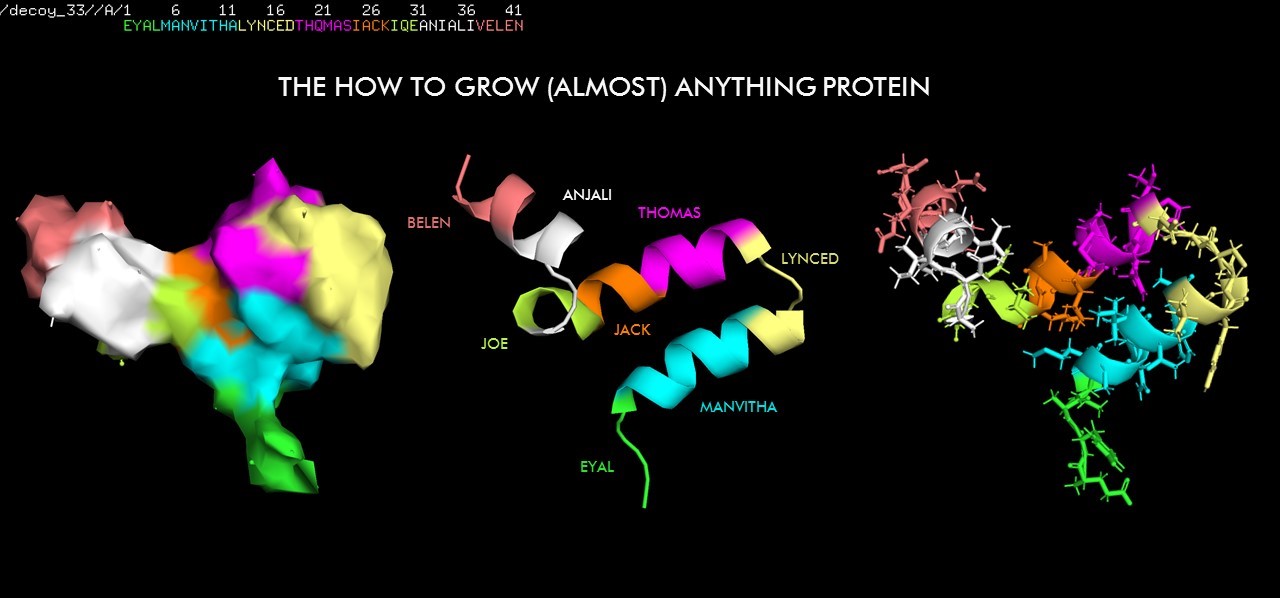
Week 4: Protein Design
Part A: Protein Analysis
1. Prelab questions
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Different types of meat have different percentage of protein, but we will assume that the average is 26%. Therefore in 500g of meat we would have 130g of proteins.
Given that 1 Dalton is 1.66054 × 10e−24 g, Therefore, there will be 130 g protein / 1.66 × 10e−24 g/Da = 7.829e+25 Da of proteins in 500g of meat.
Since each aminoacid is ~100 Daltons, then, there will be 7.829e+23 aminoacids in 500g of meat
Why are there only 20 natural amino acids?
Although the answer it is not clear, it seems to be related to their chemical properties.
Althoughtheoretically there could by 64 (4^3) aminoacids, the 20 natural aminoacids seem to have higher polymerization reactivity and fewer side reaction, which make them more stable and prone to allow life.
Why most molecular helices are right handed?
Jack Dunitz
published a paper in 2001 explaining how Pauling in his first representation of the alpha helix had predicted a left-handed structure that turned out to be wrong since the right structure was
its enantomer. As with DNA, the aminoacid helices bear a diastereomeric relationship to the chirality of the amino acids; both have to be inverted to get the proper enantiomer. I found a great
explanation in this page.
Where did amino acids come from before enzymes that make them, and before life started?
In 1953, Miller and Urey attempted to re-create the conditions of primordial Earth. In a flask, they combined ammonia, hydrogen, methane, and water vapor plus electrical sparks (Miller 1953). They found that new molecules were formed, and they identified these molecules as eleven standard amino acids.
What do digital databases and nucleosomes have in common?
Nucleosomes are the basic structural unit for DNA packaging in cells. It is forms by a segment of DNA wound around 8 histones. Databases also consist of repeated structures of information that can be retrieved when needed.
2. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions.
Briefly describe the protein you selected and why you selected it
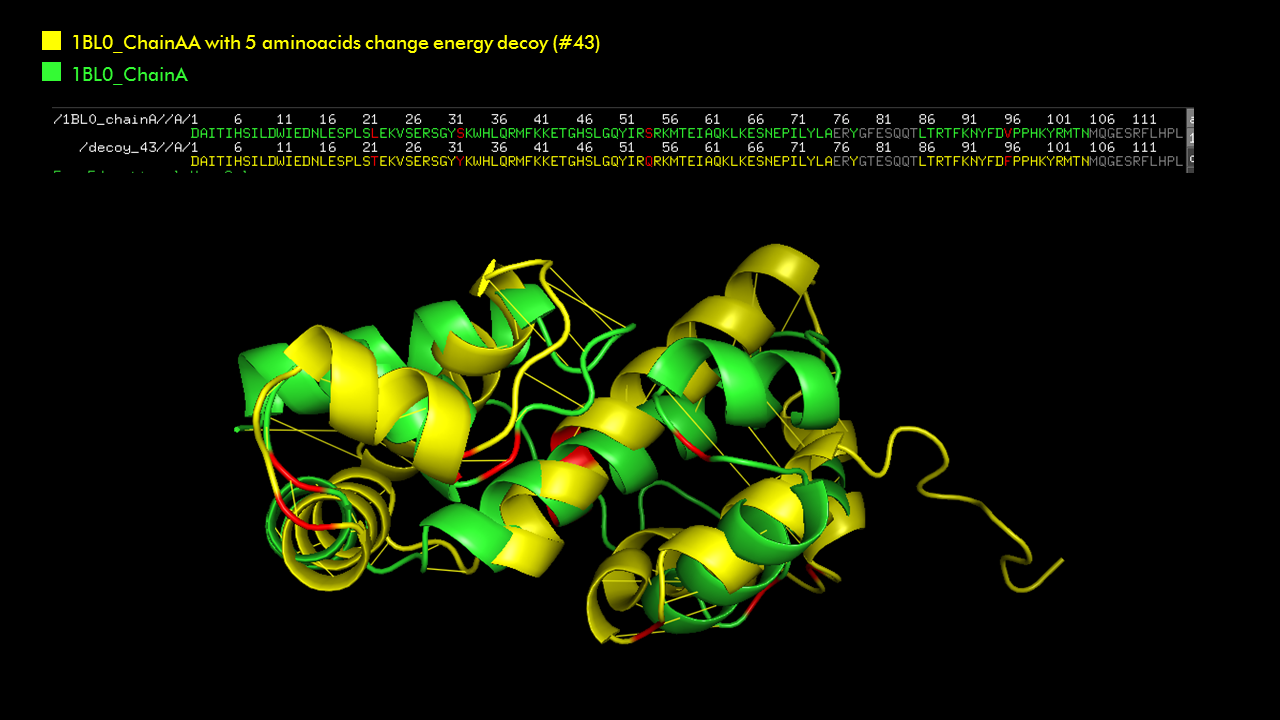
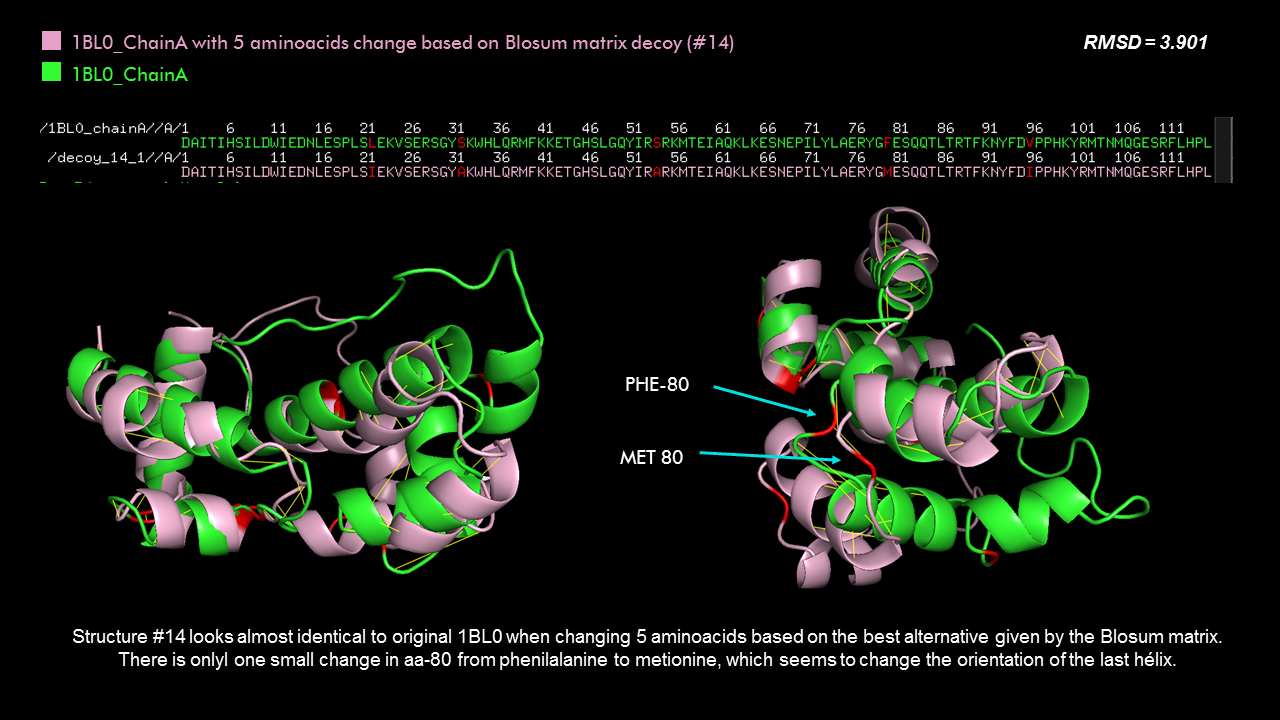
I picked 6LU7, which is the main protease of the 2019-nCoV coronavirus (also called SARS-CoV-2), that is currently posing dangers worldwide.
This protein is a dimer with 2 identical subunits (ie. 2 chains) that form 2 active sites.
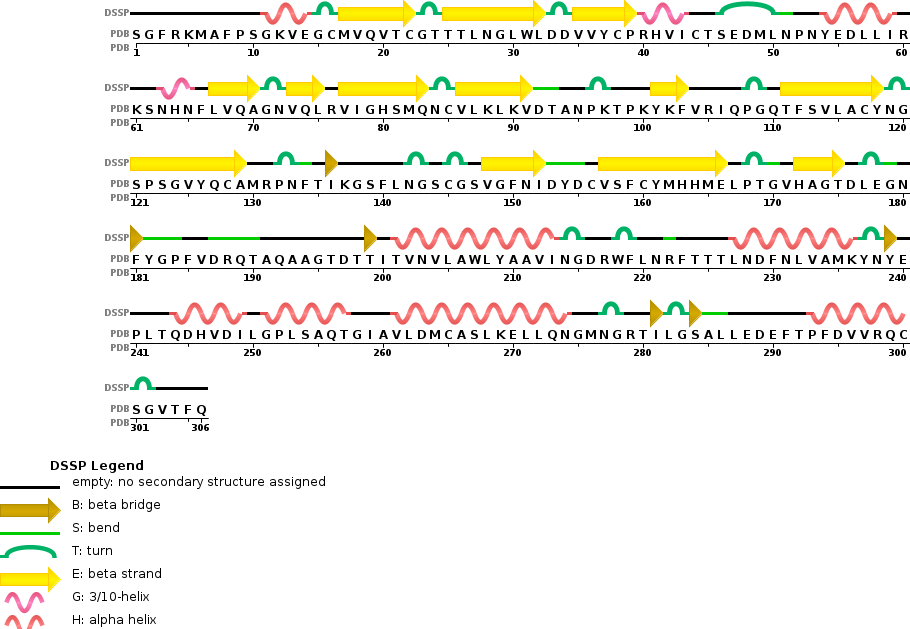
How long is it? What is the most frequent amino acid?
It has 306 residues and the most frequent one is Leucine. Below is the RSCB sequence chain view
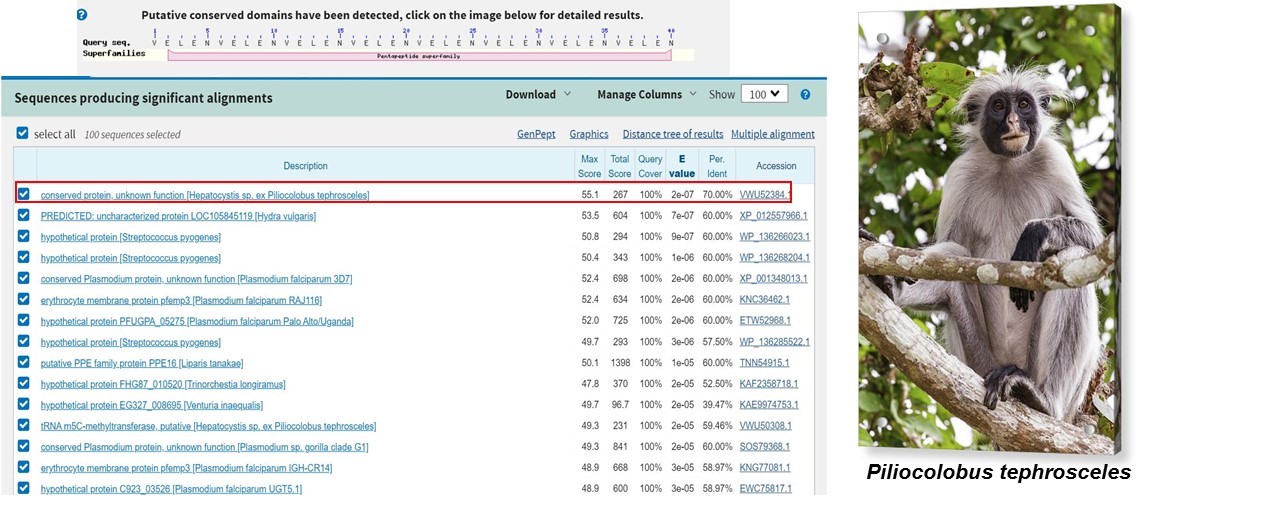
How many protein sequence homologs are there for your protein?
Using the pBLAST tool, we found more than 100 homologs in other virus such as Bat SARS-like coronavirus, Bat coronavirus RaTG13 etc.
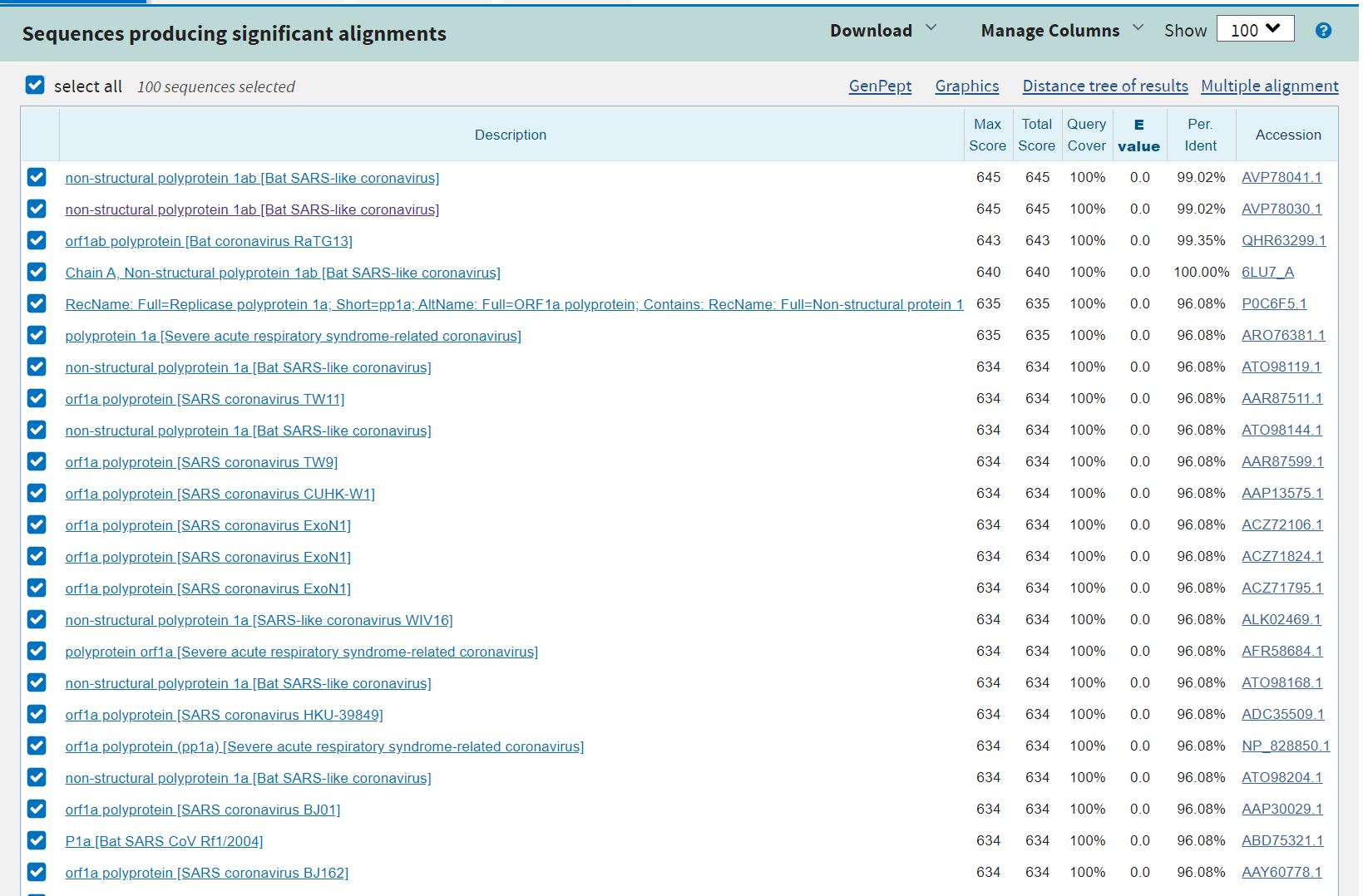
Snapshot of the results obtained in the BLAST

Snapshot of the distance tree of results
Does your protein belong to any protein family?
It is a proteinase
Identity the amino acid sequence of your protein
When was the structure solved? Is it a good quality structure?
It was deposited on January 26th 2020 and released on 2020-02-05 with a new version updated on February 26th.
Regarding the quality, here is the full report . The reported resolution of this entry is 2.16 Å.Are there any other molecules in the solved structure apart from protein?
Yes, it is studied with an inhibitor: N-[(5-METHYLISOXAZOL-3-YL)CARBONYL]ALANYL-L-VALYL-N~1~-((1R,2Z)-4-(BENZYLOXY)-4-OXO-1-{[(3R)-2-OXOPYRROLIDIN-3-YL]METHYL}BUT-2-ENYL)-L-LEUCINAMIDEDoes your protein belong to any structure classification family?
It is only classified as viral protein. Structure classification family is not yet available since it was published very recently
Identify the structure page of your protein in RCSB
Visualize the protein as "cartoon", "ribbon" and "ball and stick".
I decided to represent the biological unit of the protein, which is formed by 2 chains.
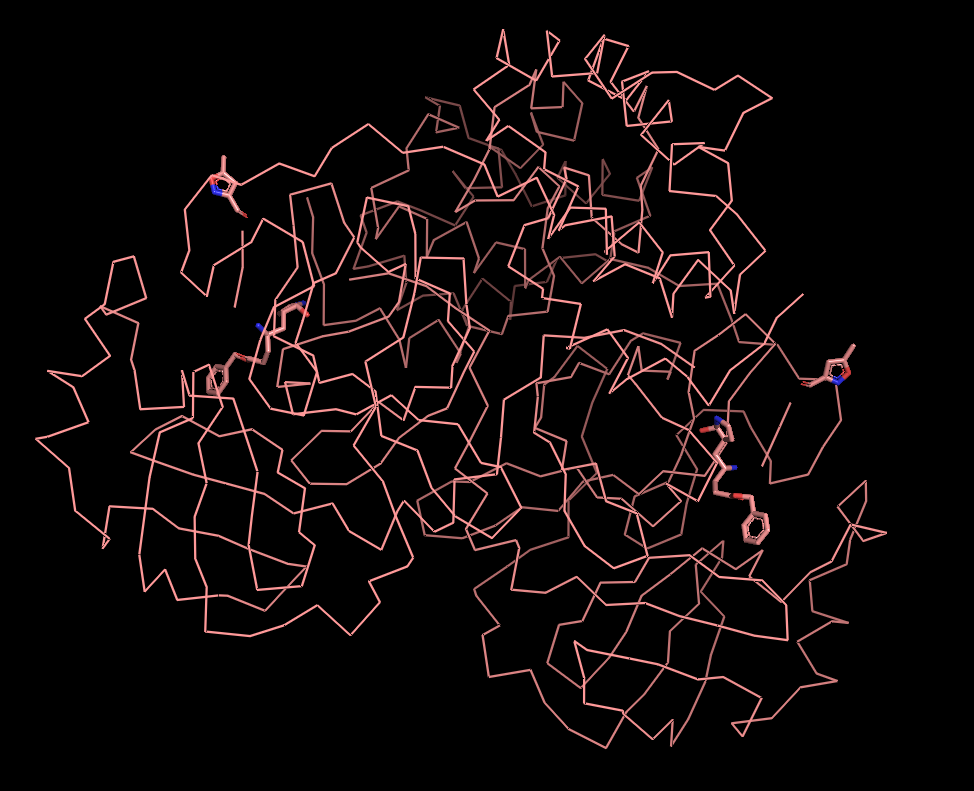
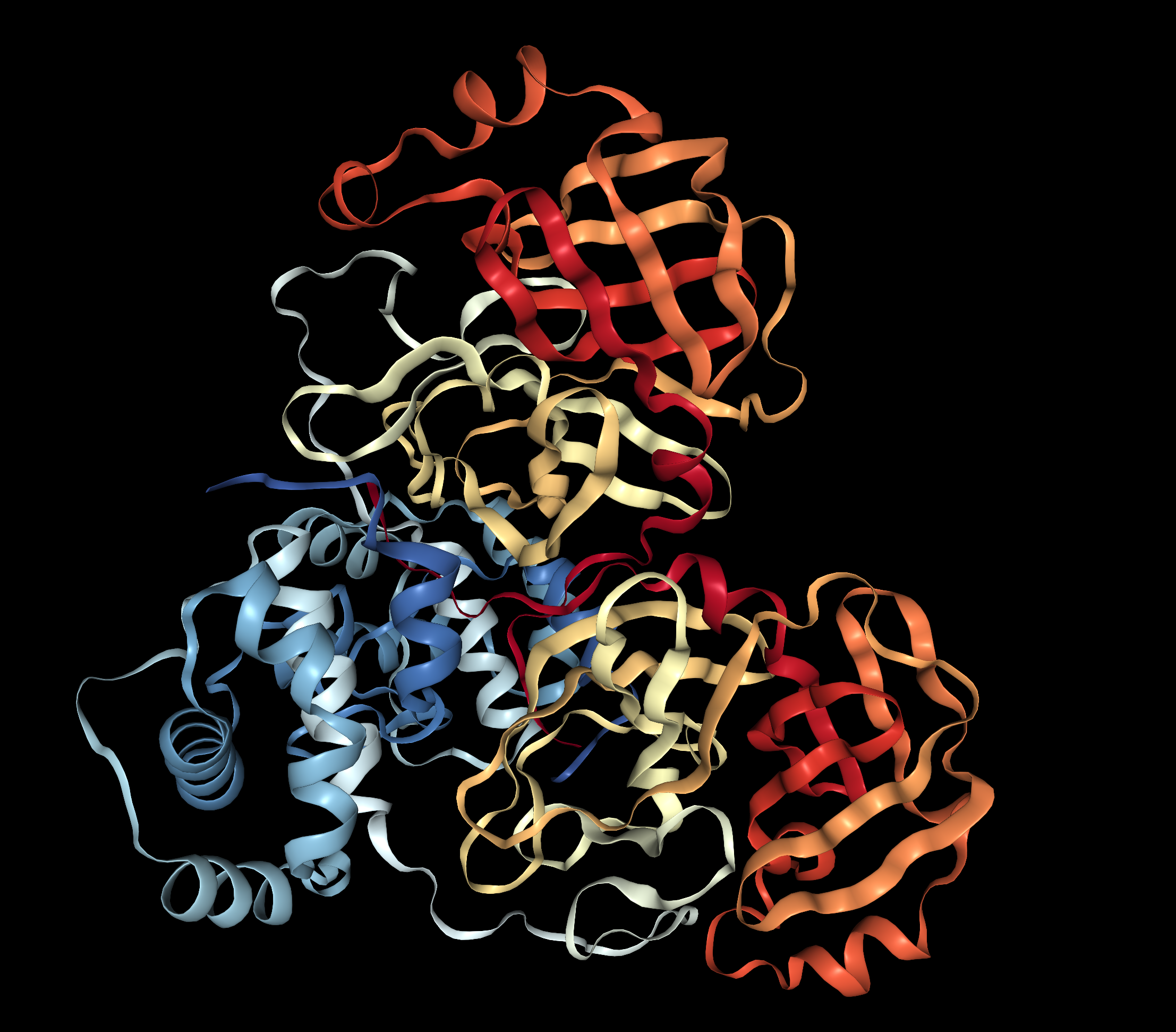

Ribbon Cartoon ; Ball and stickColor the protein by secondary structure. Does it have more helices or sheets?
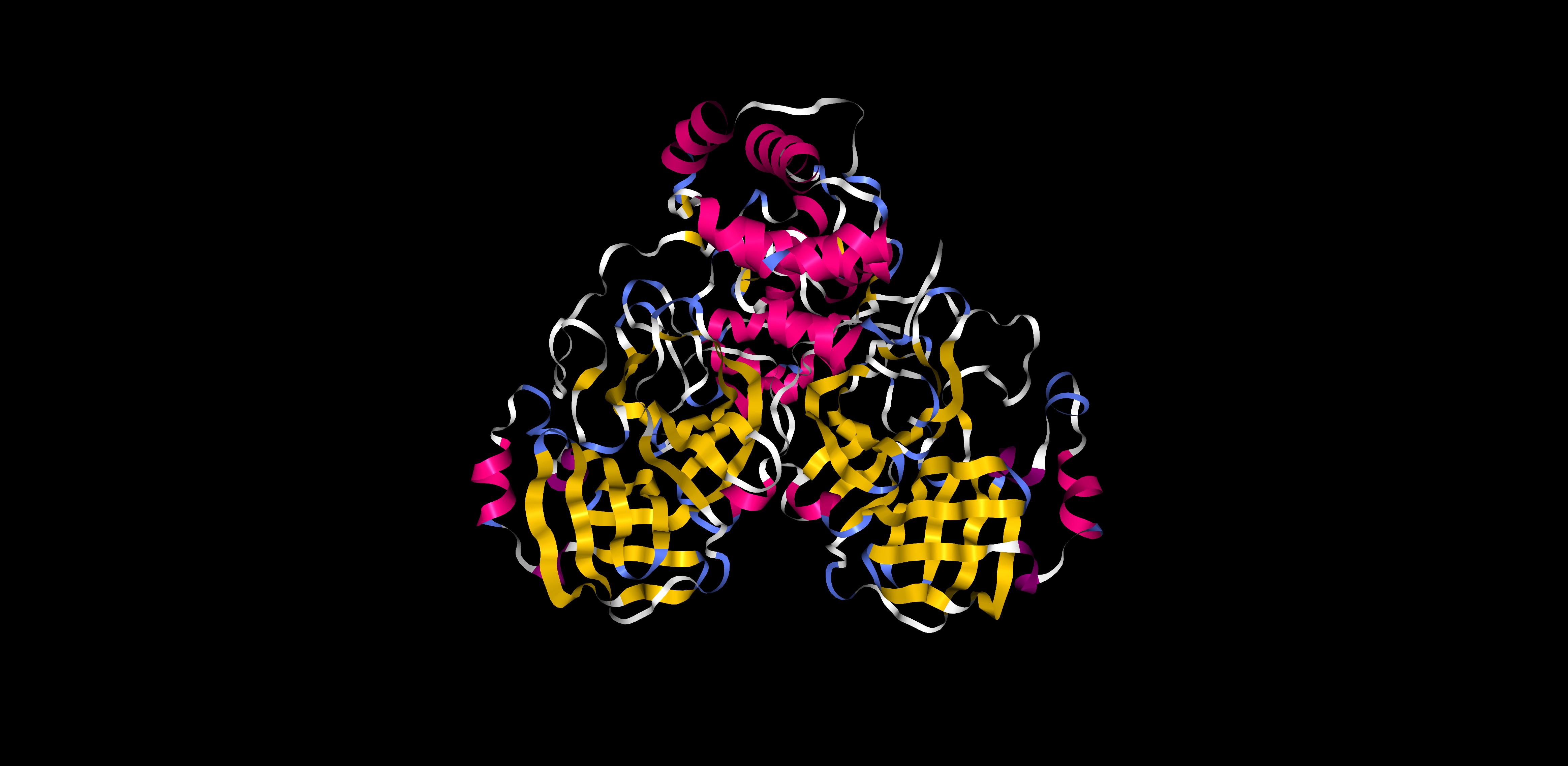
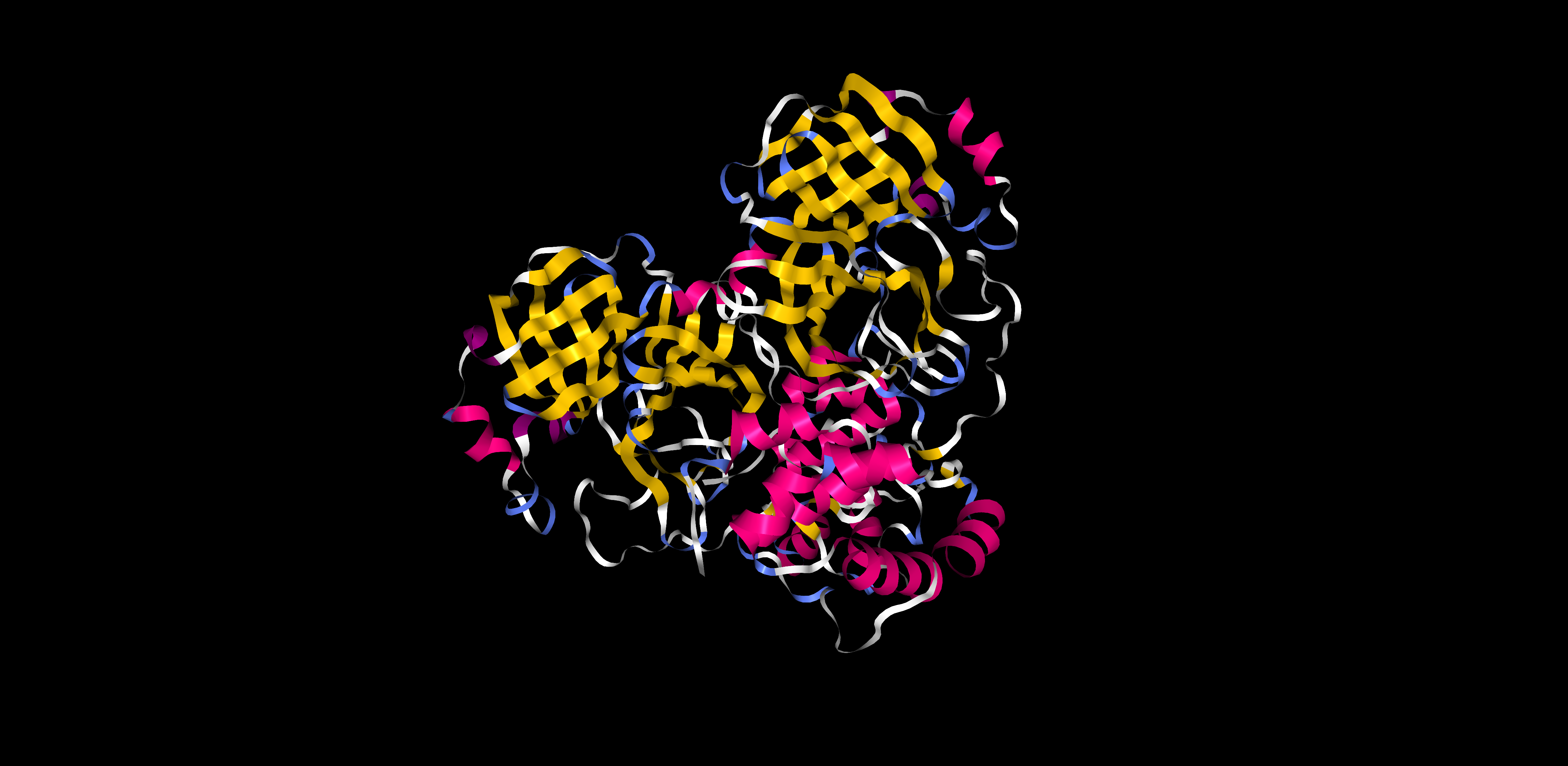
There is similar amount of helices and sheets
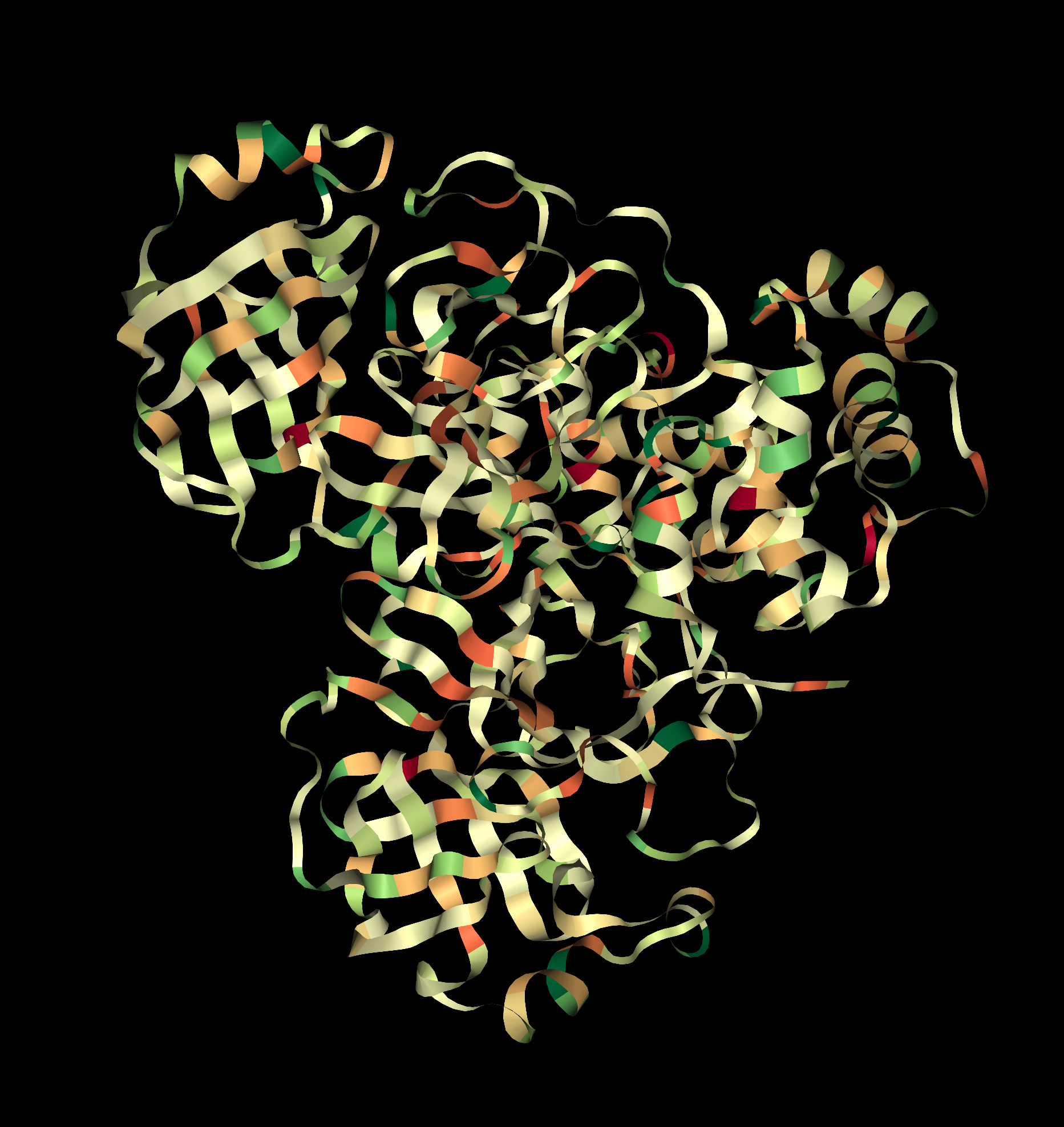
Pink: alpha helix Yellow: beta strands White: coil Blue: beta turnsColor the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
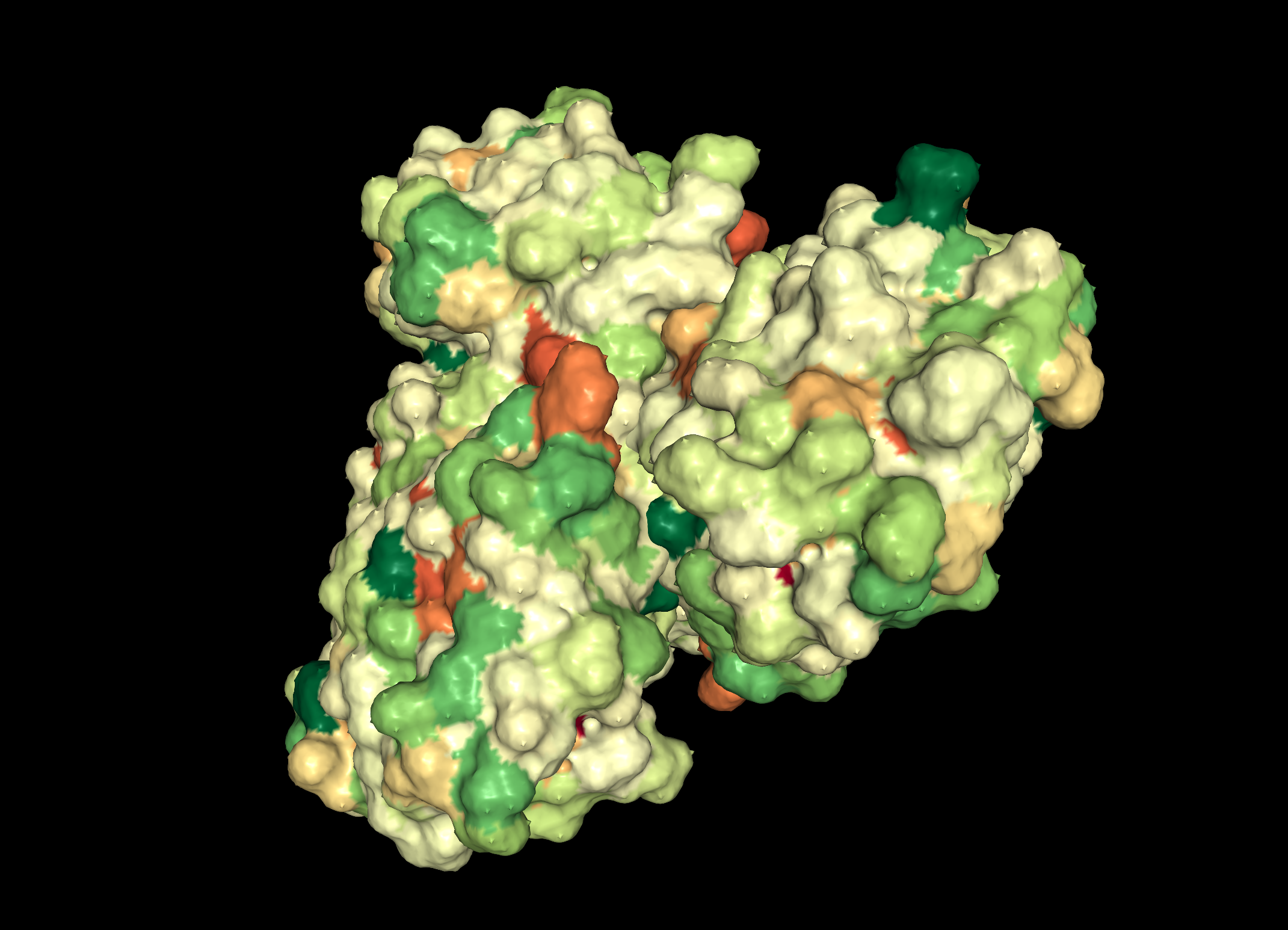
Visualize the surface of the protein. Does it have any "holes" (aka binding pockets)?
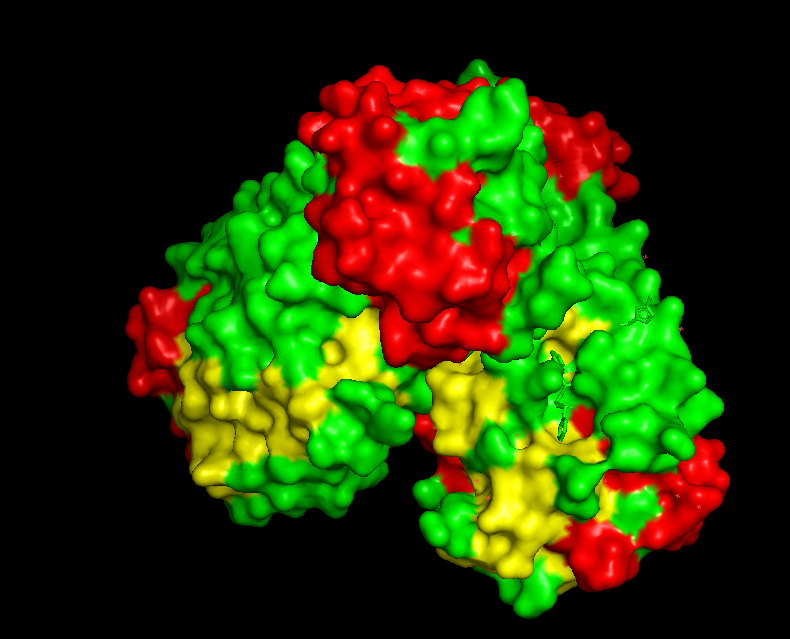
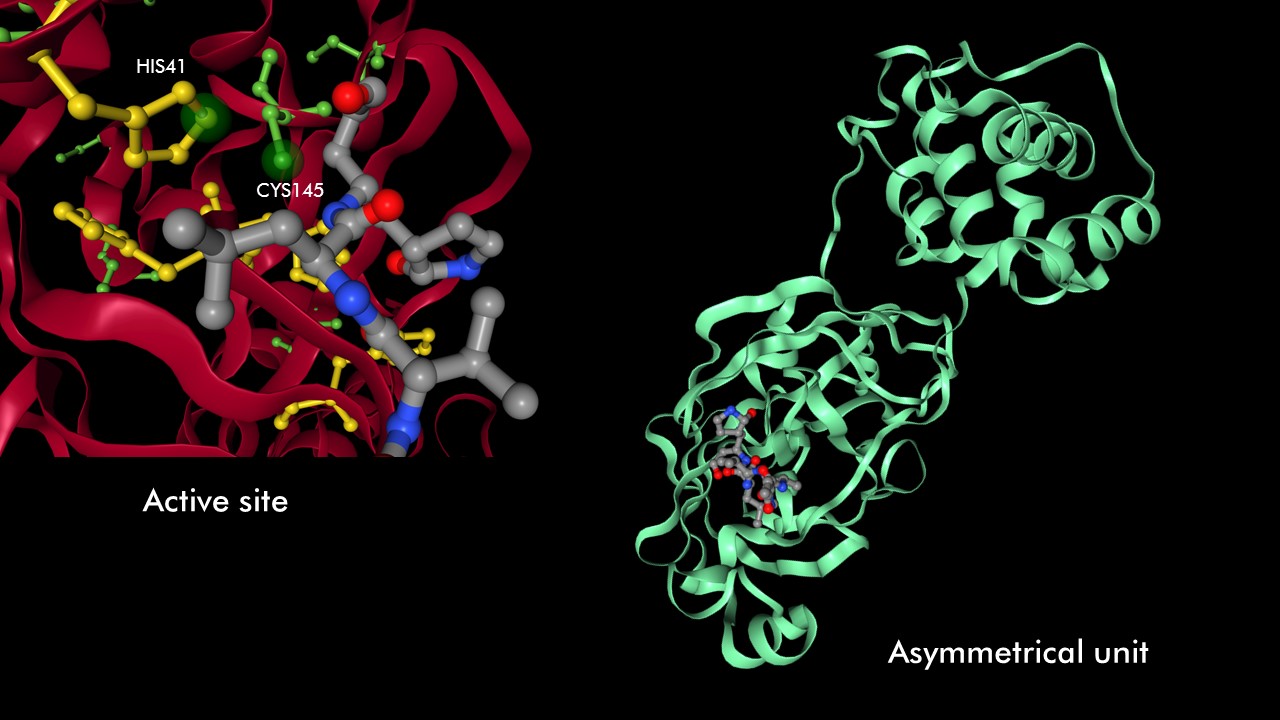
A cysteine amino acid and a nearby histidine perform the protein-cutting reaction. This structure has a peptide-like inhibitor bound in the active site
Open the structure of your protein in any 3D molecule visualization software
Proteins can be represented as assymetric unit, biological unit, unitcell and supercell. For 6LU7, we will use the biological unit in all representations, since that is considered the functional subunit (2 chains)
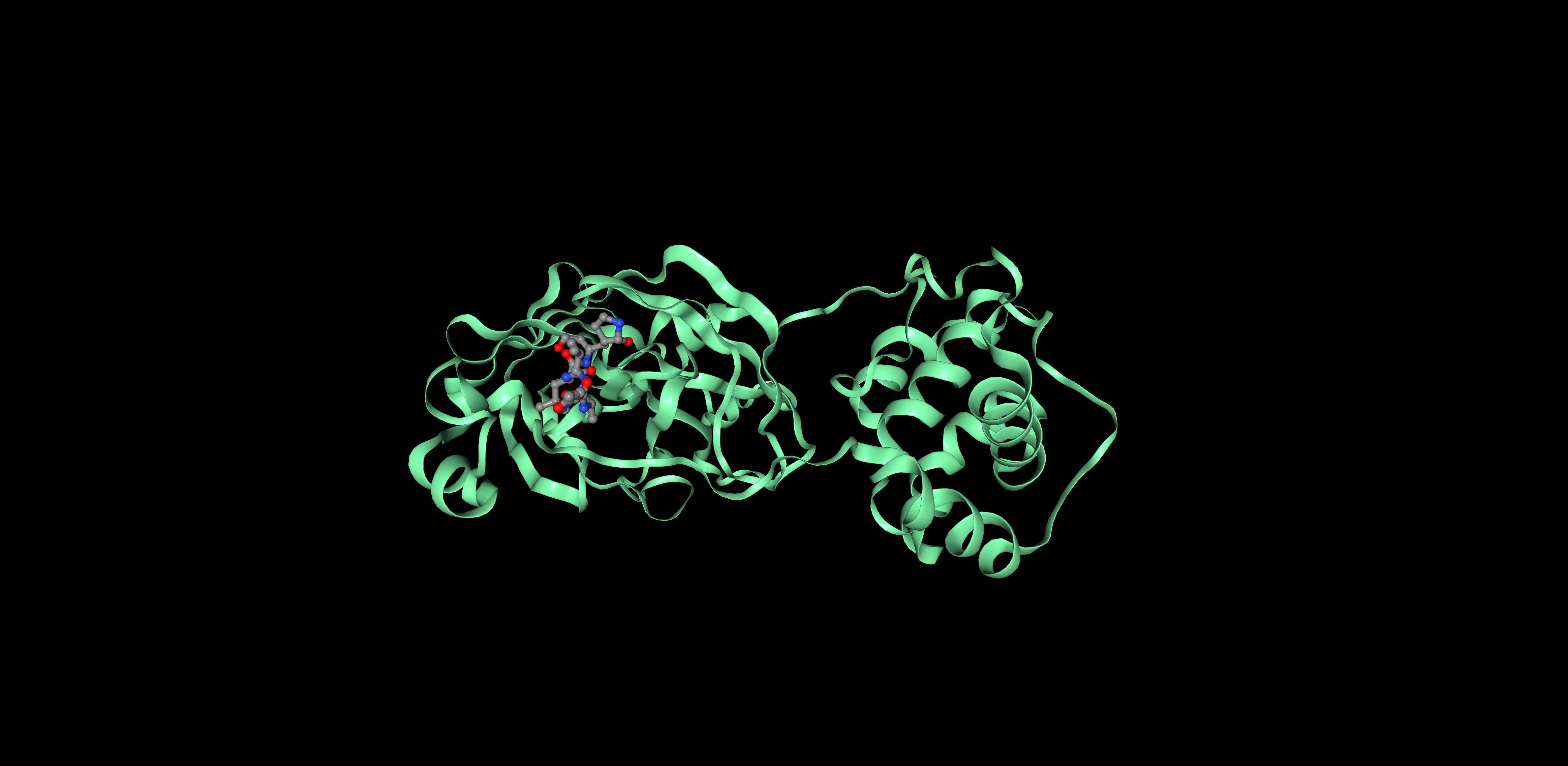
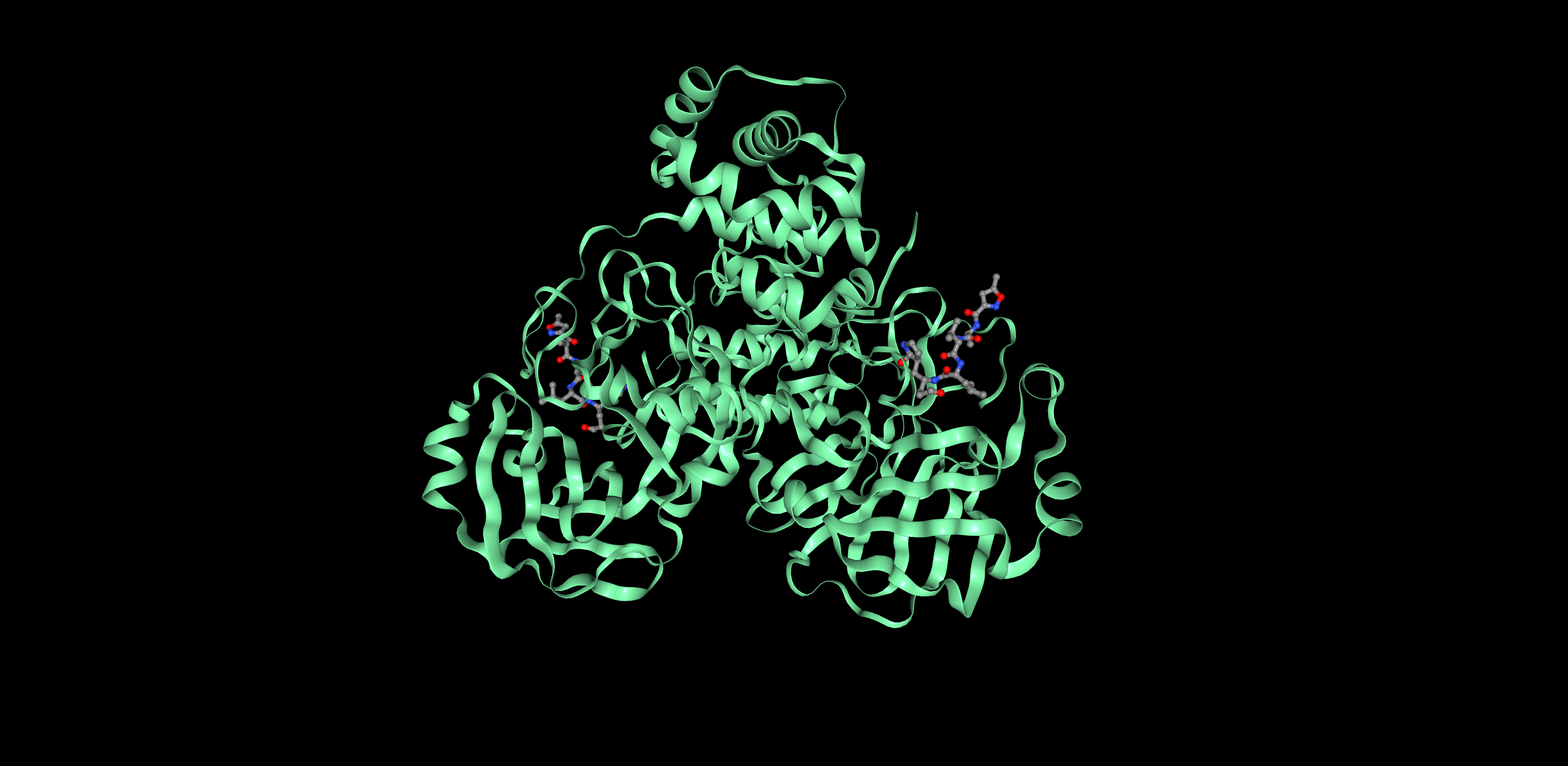
Assymetric Unit Biological unit
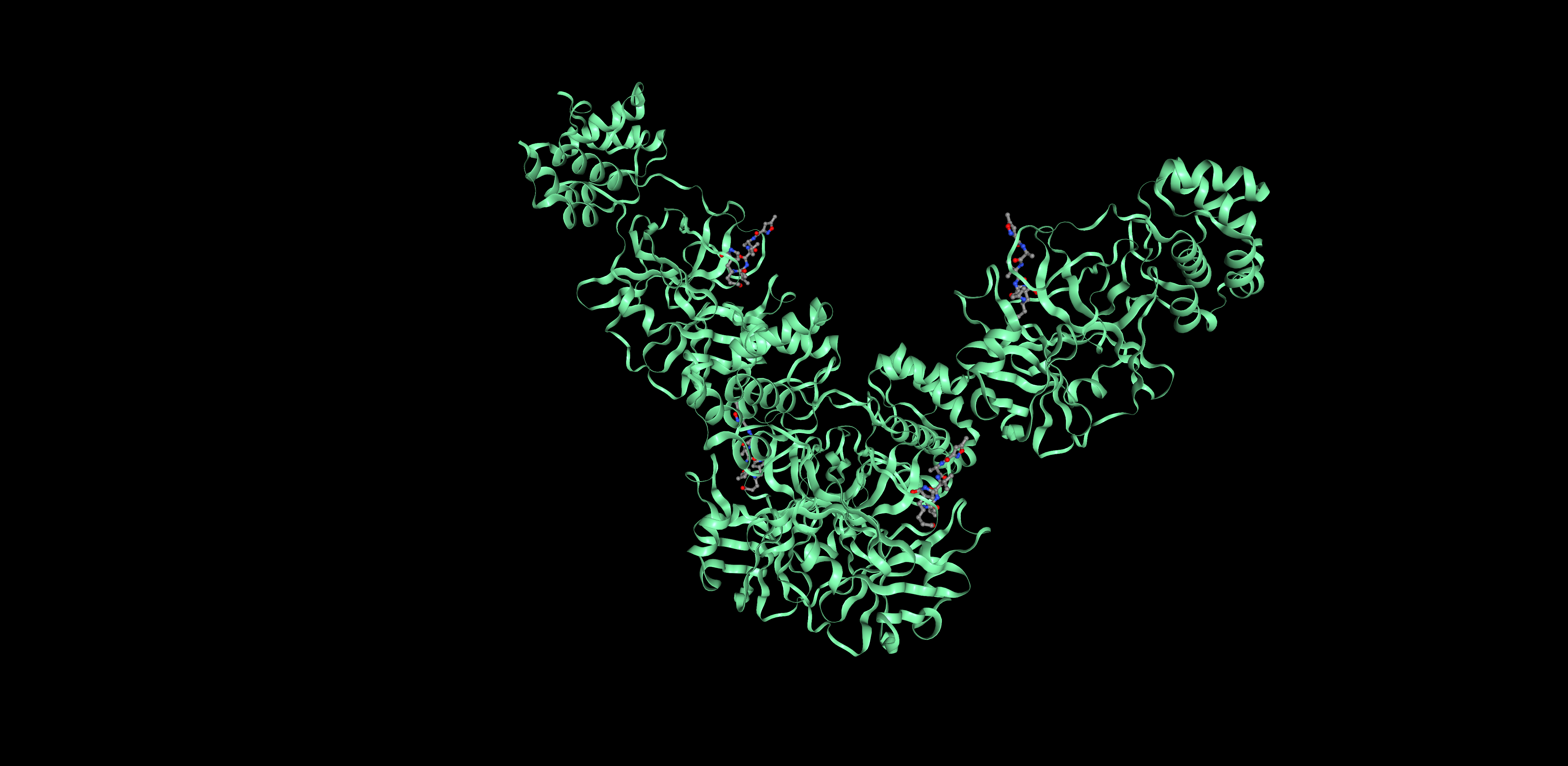

Unit cell Super cell