Week 3 - Protein Design
Part A: Protein Analysis
General Questions
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
- A Dalton is also know as an atomic mass unit, it expresses weight so it can easily be converted into grams [1].
- A good average of percentage of proteins in meet is ~22% [2] => that means that we have (500 * .22) grams of protein = 110g .
- 1 Dalton = 1.6605e-24 grams and an aminoacid ~100 Daltons => We have around (100 x 1.6605e-24) = ~1.66e-22 grams of aminoacids in our 500 gram meat.
- So that would mean that we have (110 / 1.66e-22) ~6.62e23 aminoacids in our piece of meet. Wow, thats a lot of aminoacids!!!
- Why are there only 20 natural amino acids?
Aminoacids are created through codons. Codons are a chain of three bases stuck together so that would mean that there are a total of 4^3 (64) possible combinations of aminoacids. Nature has evolved to produce only 20 in part because of inherited redundancy, this means that there are several codons that encode for the same aminoacid. This makes encoding more robust and high fidelity translations. Mutations are also harder when you have this type of redundancy [3]. Nature is wise!
- Why most molecular helices are right handed?
Theoretically, the bonds can be made as to form both right handed and left handed helices. In the real world, right handed helices is energetically more stable, meaning that it has a lower state of energy due to having fewer steric clashes between the side chain andf the main chain [4].
- Where did amino acids come from before enzymes that make them, and before life started?
In the origins of life on Earth, aminoacids were synthetized chemically from a large mixture of organic compounds. After more complex forms of life arose, the ones that were able to synthesize their own aminoacids survived and thats why, today, organisms are all able to synthetize their own aminos through enzymes and methabolic pathways [5]. The image from nature education below shows the history of how these molecules were sytethized in nature [5].
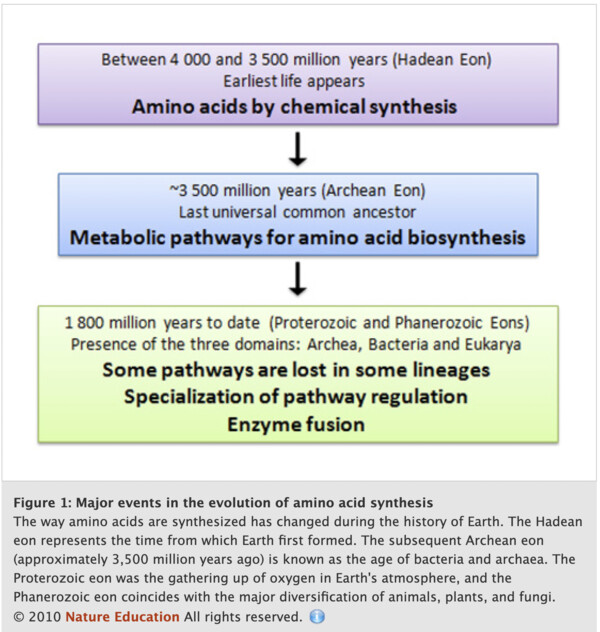
- What do digital databases and nucleosomes have in common?
Nucleosomes are proteins that help to wrap DNA into small structures so that it can fit within a nucleus. They are similar to digital databases because they are what enables cells and organisms to pack a lot of information within cells [6].
- Briefly describe the protein you selected and why you selected it.
I chose to analyze the TlpA protein present in Salmonella. In electronics, one of the most simple sensors is the temperature sensor, therefore I would love to explore the possibility of creating my bio temperature sensor. This protein acts as a transcriptional repressor. The protein dislocates from DNA with temperature shock allowing the repressed transcriptions to occur.
- Description of amino acid sequence.
Length: 371 - Most Frequent Amino: A/Alanine (57)
Protein Sequence Homologus:
I used BLASTP to see the sequences that produce significant alignment with my protein and found around 100 of them. Nevertheless, many of them seem to be showing the exact same sequence and name.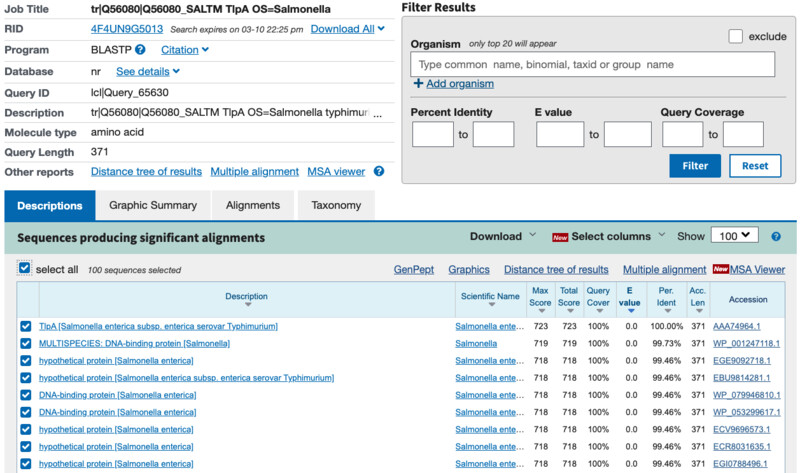 Protein Family:
Protein Family:
Looking into the family and domain infroamtion on uniport I see that Its part of the KfrA_DNA-bd_N family of proteins.
- Protein Structure on RCSB.
Structure Solved Date.
I was unable to find my exact same protein structure on RCSB but I was able to find it on the SMR (Swiss Model Repository). The structure seems to have been solved 2017-09-07 and seems to be well annotated and docuemnted. I was unable to find any structure classification family information.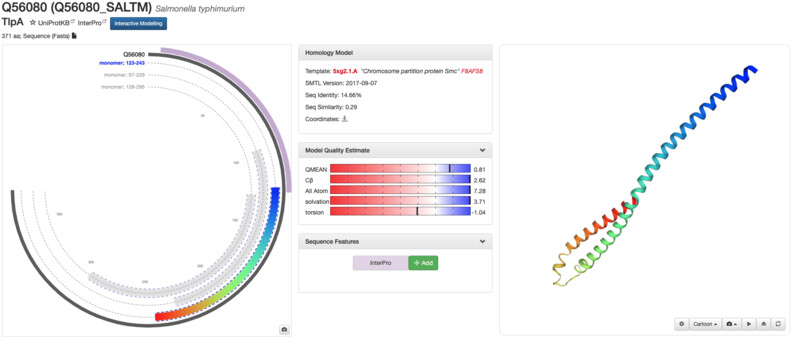
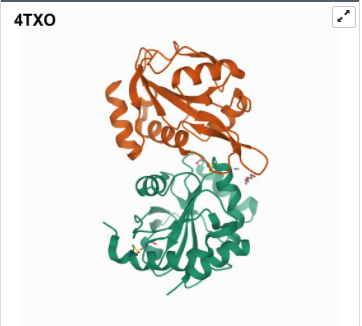
I was a bit skeptical that the structure seemed to be very simple and looked it up agin in the on RCSB, they showed some more complex results for proteins that seem to be TlpA like. Here are some of the images.
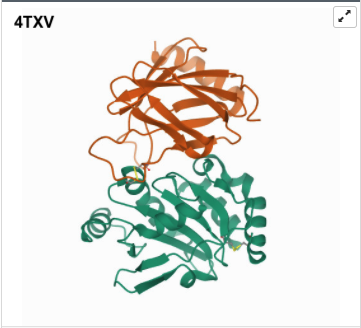
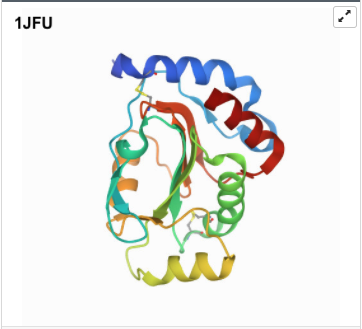
- 3D Protein Visualization.
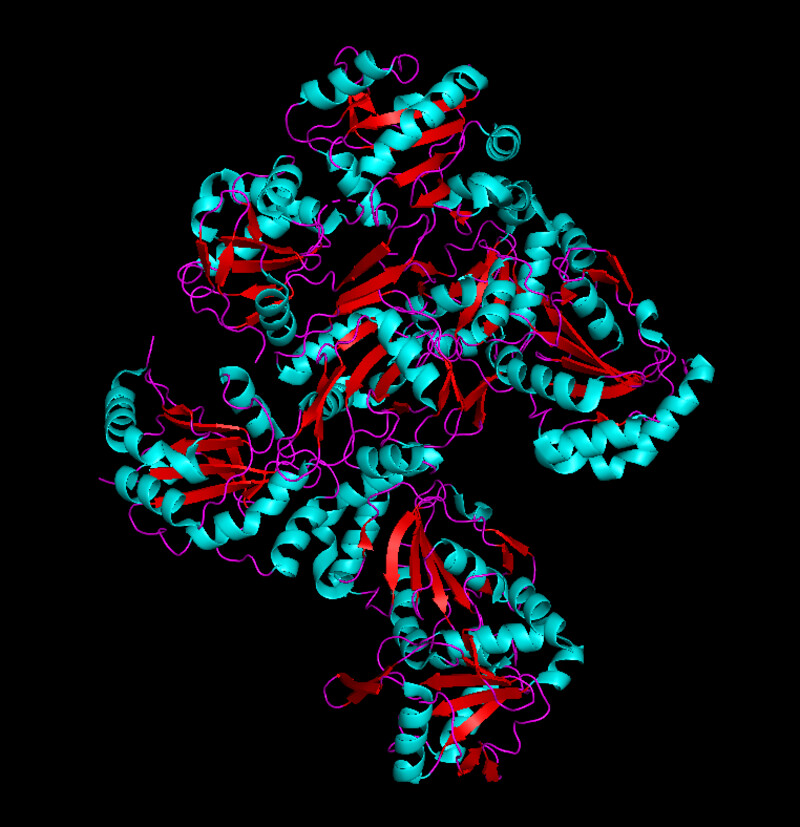
I was able to use the SMR 3D visualization tool to further analyze and play around with my chosen protein. Below are the different views it has.

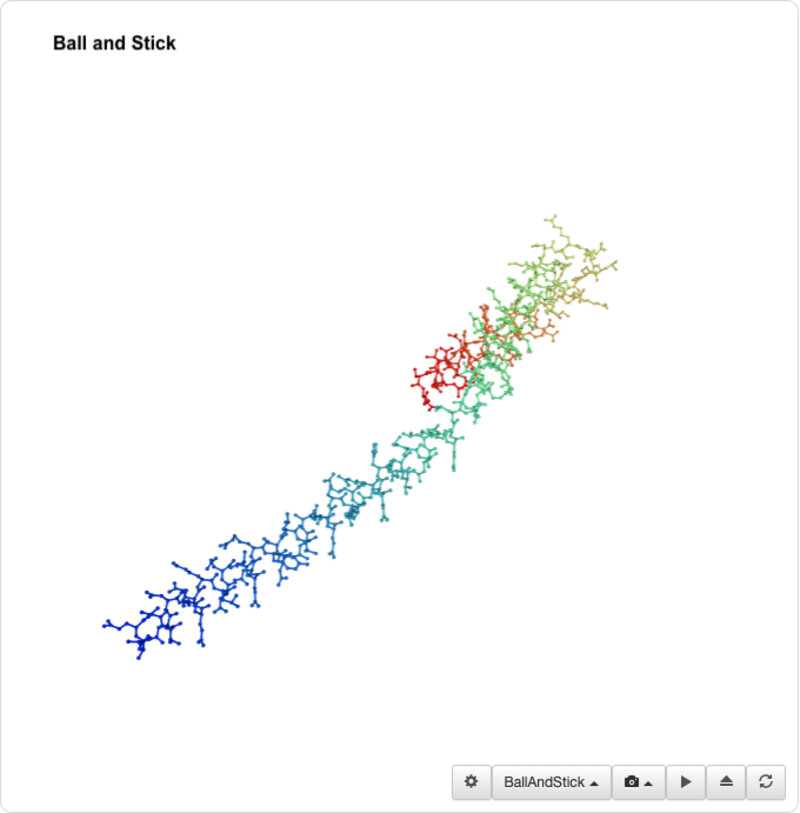

I was unable to find a proper pdb file for the protein that I've been using so I decided to use one of the similar ones shown above (4txo) so that I coul learn how to use the functions from PyMol. I show the new views of the structure below:
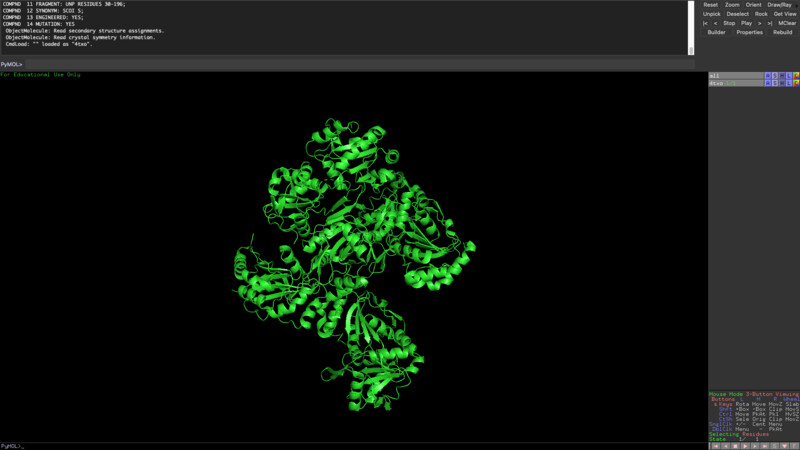
The new sequence seems to have more alpha helixes but it still has quite a lot of beta sheets as opposed to my initial protein visualization.
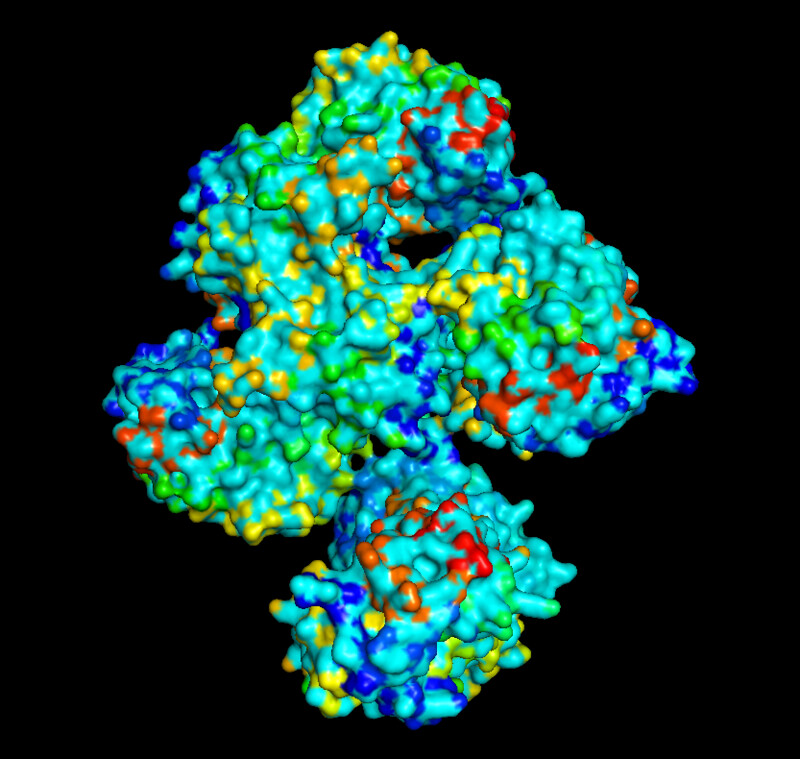
 From analyzing the surface I would also conclude that the protein does have several 'holes' or binding sites.
From analyzing the surface I would also conclude that the protein does have several 'holes' or binding sites.
Part B: How to (almost) Fold (almost) Anything
I went on to the second part of the asignment. For this section, I looked for a Protein that had less than 100AA. I chose the common Leucine zipper. The protein is able to bind at specific promoter sites and stimulate expression of specific genes. This is a motif which means that it can be found as a section or region of a larger protein. After choosing the protein, I downloaded its .pbd and .fast files from RCSB and submitted the .fast file for folding online on robetta.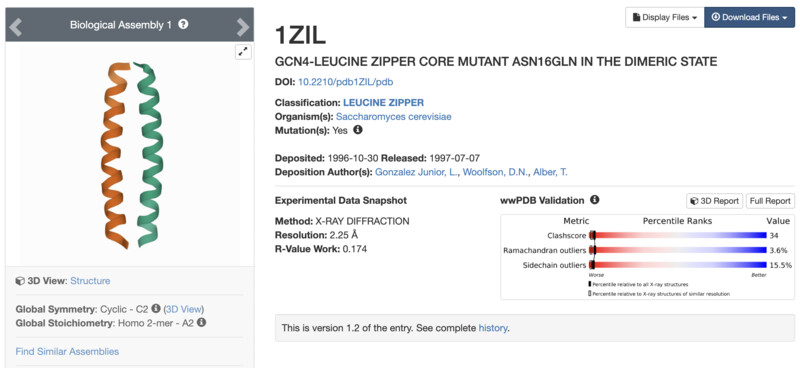
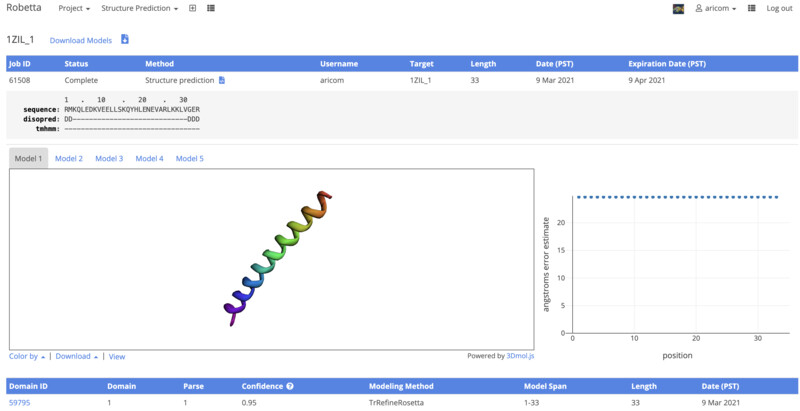
I received an email from Robetta a day after my submission. I logged into find the results. From the visualization given by robetta I can see that it its very similar to the one that I was shown in pyMol before. There is only one half of my entire structure, I belive that is due to the sequence being the same for both halves. Next, I will align the results on pyMol.
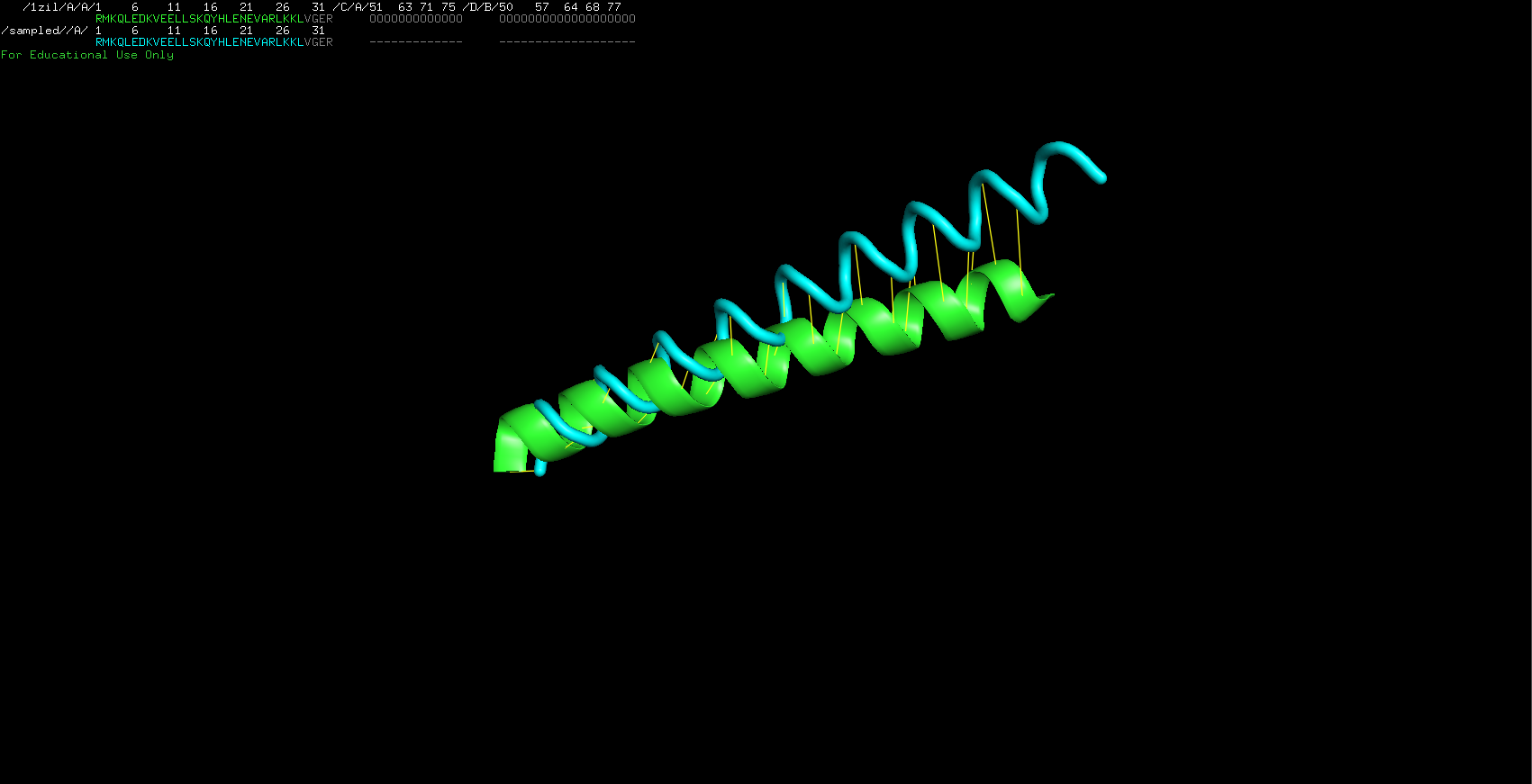
I imported both of the pbd models into PyMol. They look very similar in shape but are not perfectly identical. I then used the alignment tool built into python and ontain the following result. The alignment is not perfect indeed but it does not show too much of a difference (illustrated by the yellow lines connecting the molecules). The downloaded file is represented in green and my robetta folded file is in blue.
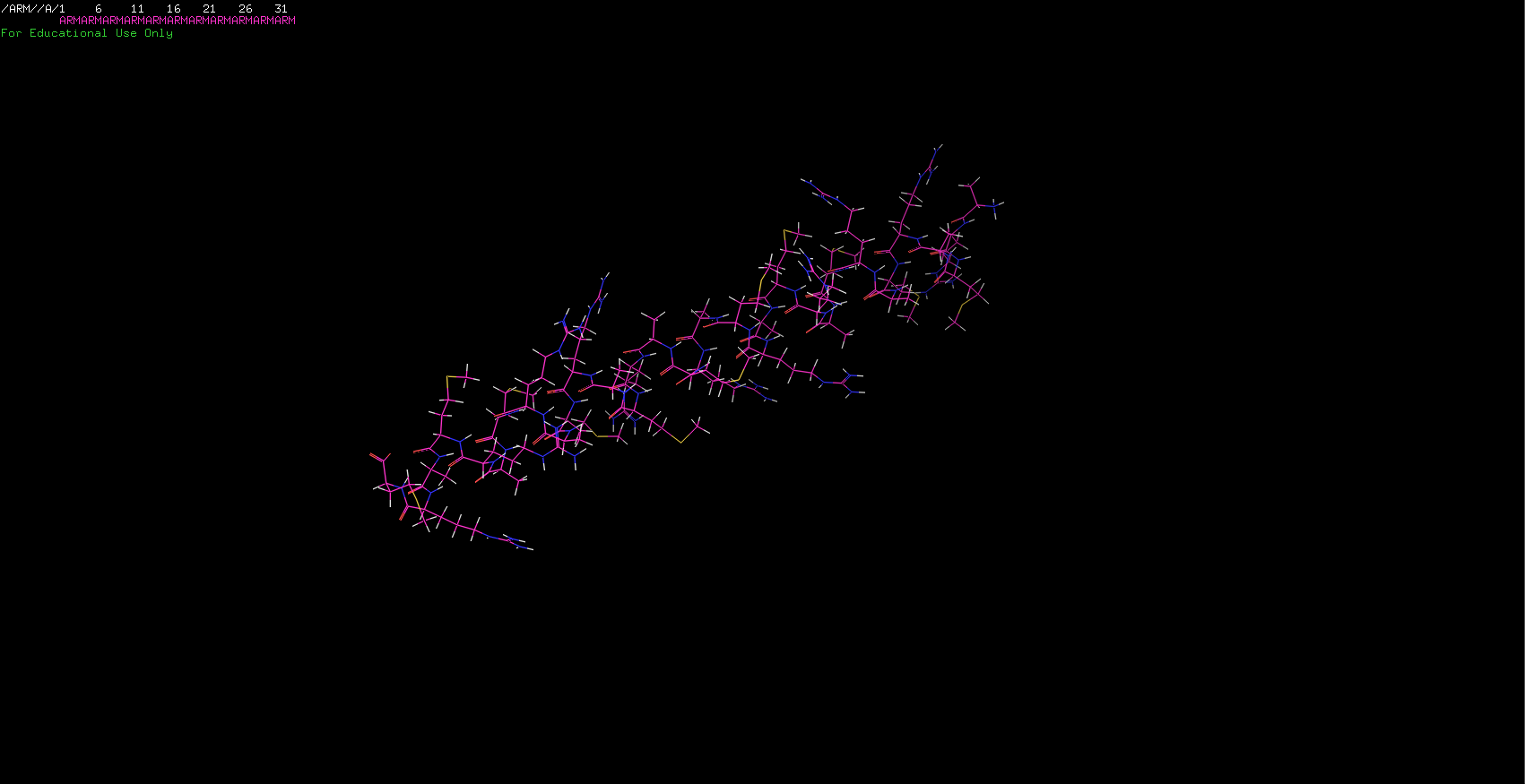
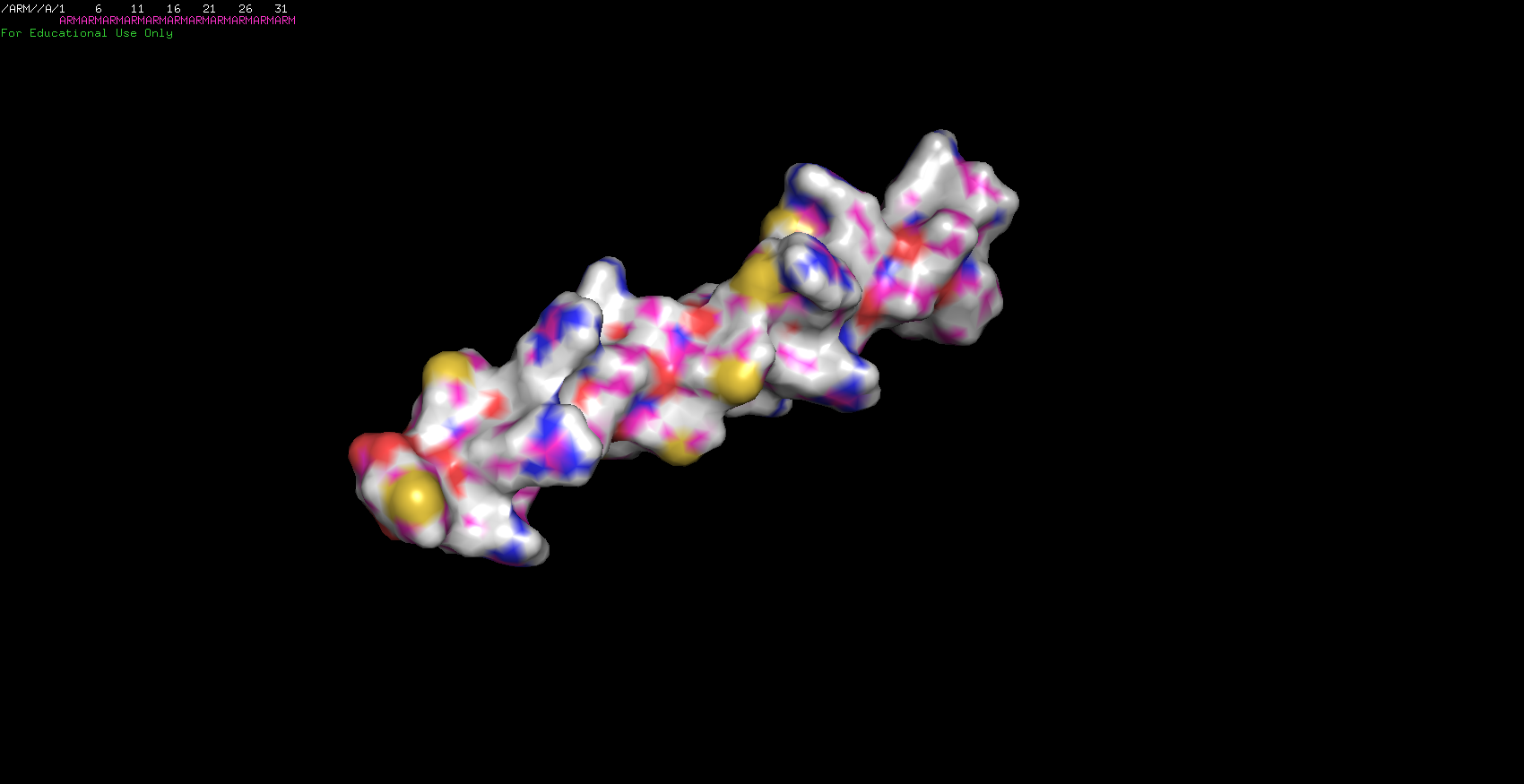
I sent out to fold two more sequences. The first one was my initials stringed together. Surprisingly, it worked! Since I was only using three aminos I was expecting to have a simple molecule and was right about it. The ARM protein is just a nice little alpha-helix!
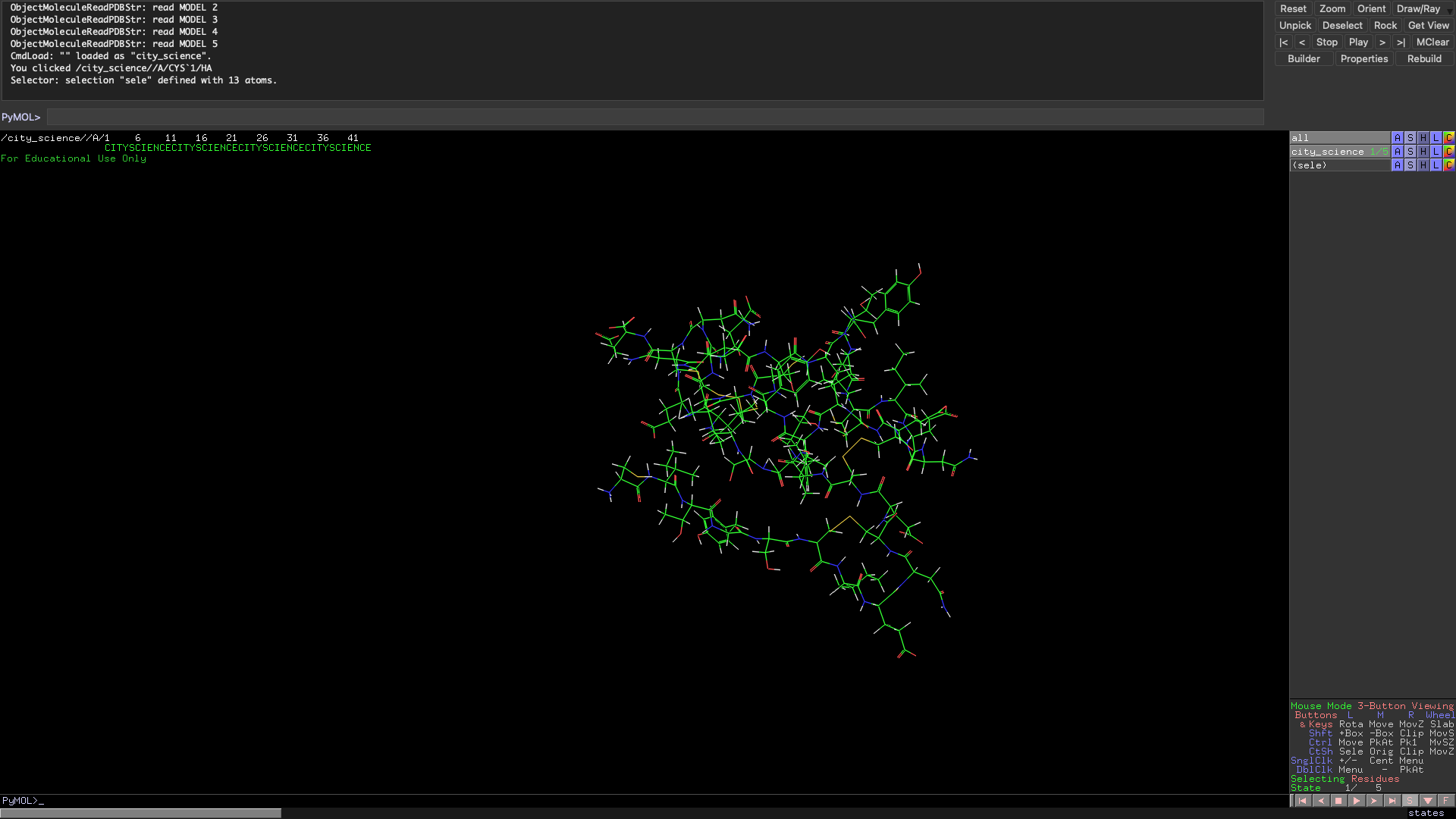
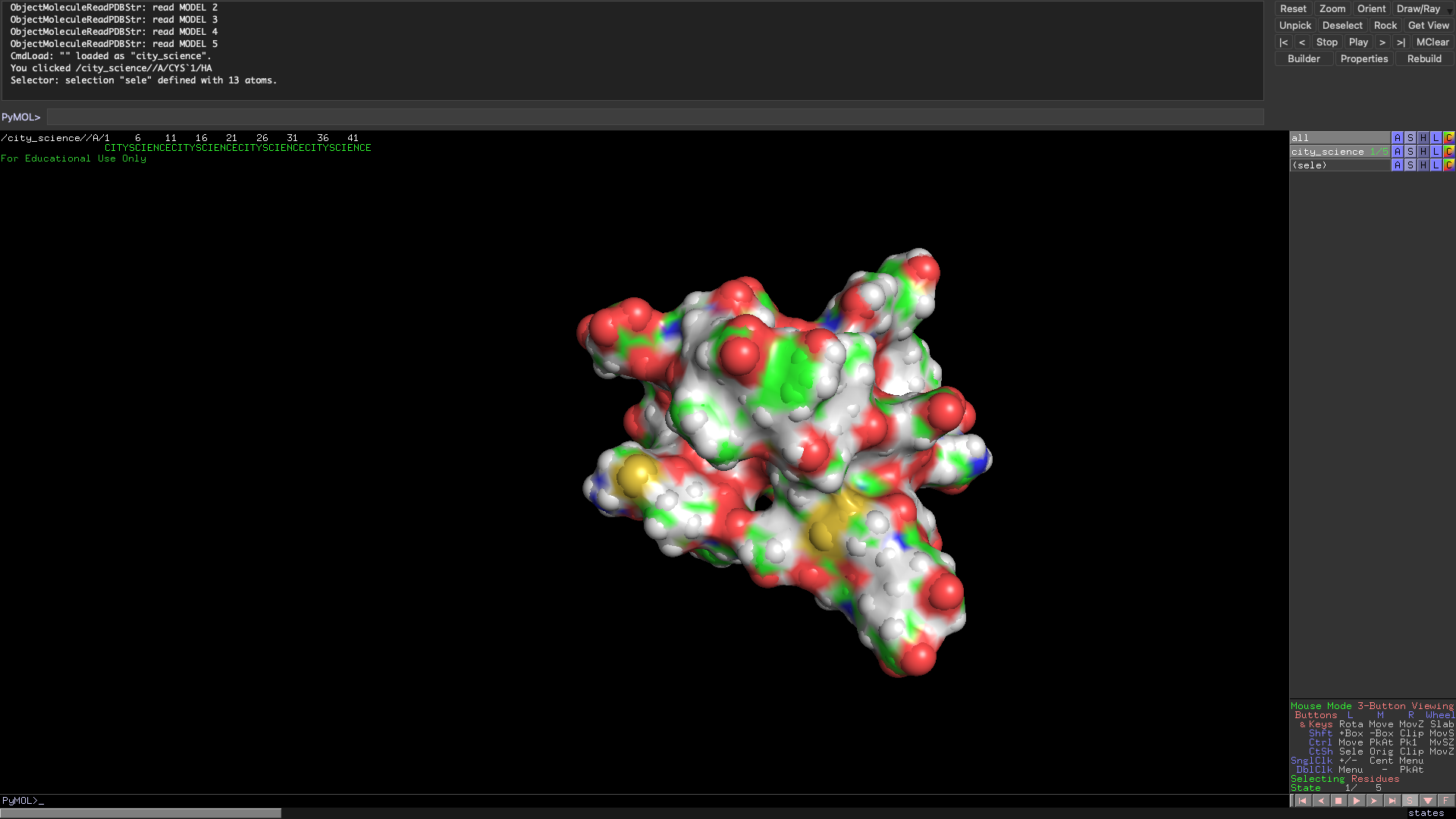
In the spirit of the Media Lab, I have also folded a City Science molecule. The group does not usually do synthetic biology but now that we have our own protein I think we will start to head into biological paths soon! The Scity Scicence sequence has more residues than the ARM so it is reasonable that the folding patter was much more complex.
Local Protein Folding Using Rosetta.
I went on to install PyRosetta locally on my machine. I'm running python3.8 on a mac. I was able to install pyrosetta by downloading my corresponding .whl file from here After I just had to navigate to the download path from my terminal and run a simple pip3 install {pyrosetta}.whl. I prefer scripting over Jupyter notebooks so I built my own script based on the Jupyter notebook. The first section of the script gave me the output below, which meant that my installation was succesful and that I was properly reading my .pbd file corresponding to my protein of intrest.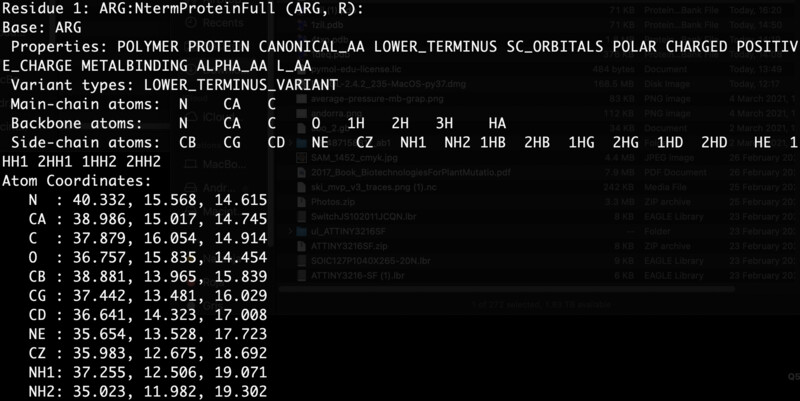
Part C: Protein Design by Machine Learning
For the last part of the assignment we looked into using AI to start identifying or improving certain protein designs so that we can control and enhance some of their characteristics and main functionalities. To do this, synthetic biology has borrowed the use of embedings from the fiel of Natural Language Processing. An embedding is the transformation of one type of data into information that can be mapped out into other useful dinmension thereby helping with knowledge abstraction from a given set of data poitns. In NLP embeddings are used to understand language, in synthetic biolofy this techniques can be helpful for understanding and comparing protein properties and functionalities.The code is for the excersise was supplied by the HTGAA teaching team and my copy of it can be found here. I ran the complete notebook and tested the three regressor types that we could choose (K-nearest-neighbors / SVM / Random Forest Regression). Below are my results:
- K-Nearest-Neighbors: SpearmanrResult(correlation=0.7764823981001997, pvalue=1.9385892992170437e-218)
- Supprt Vector Machine (SVM): SpearmanrResult(correlation=0.7803063886153849, pvalue=5.613981304588845e-222)
- Random Forest Regression: SpearmanrResult(correlation=0.7211576393257717, pvalue=4.817545694209681e-174)
Refrences:
[1][2]
[3]
[4]
[5]
[6]