Next Generation Synthesis: Gibson Assembly
March 7, 2019
New advances in biotechnology have enabled us to generate novel DNA quickly. While we're still far from creating an entire genome de novo, modifying genes is relatively easy. This week, we modified amilCP, a chromophore from the coral Acropora millepora, and inserted it into E. coli using Gibson assembly and primer defined PCR deletion.
To construct the plasmids, we used Gibson assembly. This works by using PCR primers that are half from the plasmid with the amilCP gene and half from the pUC19 sequence prior to the PvuII restriction cut site. Because of this, PCR is able to amplify the amilCP gene with an additional sequence at each end of the gene to enable it to be inserted into the pUC19 genome. In order to mutate the amilCP gene, the gene is actually amplified in 2 separate sections, with PCR primers in the middle of the gene. These primers are slightly mismatched in order to create the mutation.
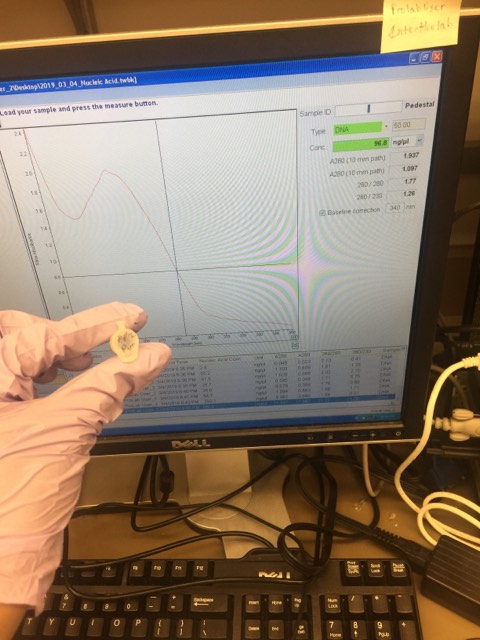
We used .5 µM of each primer in our 50 µL reaction, so that means we had 50 pmol of primer. We had 200 ng of DNA of a 2924 bp plasmid. By the NEBio calculator, we had 221.4 fmol of plasmid. Dividing the amount of primer by the amount of plasmid, we would need 225 cycles to use up all of the primer.
The Gibson assembly mix contains 3 different enzymes to combine the different DNA pieces. Exonuclease removes some of the base pairs from the 5' strand on each end. This allows the pieces with complementary ends to anneal together. DNA Polymerase is then used to fill in any gaps left by the exonuclease, and DNA ligase glues together the filled in section.
The overall Gibson Assembly reaction is incubated at 50°C, which is slightly less than the melting temperatures for the overlaps (between 53 and 54°C). This enables them to anneal at the fastest rate possible without breaking apart.

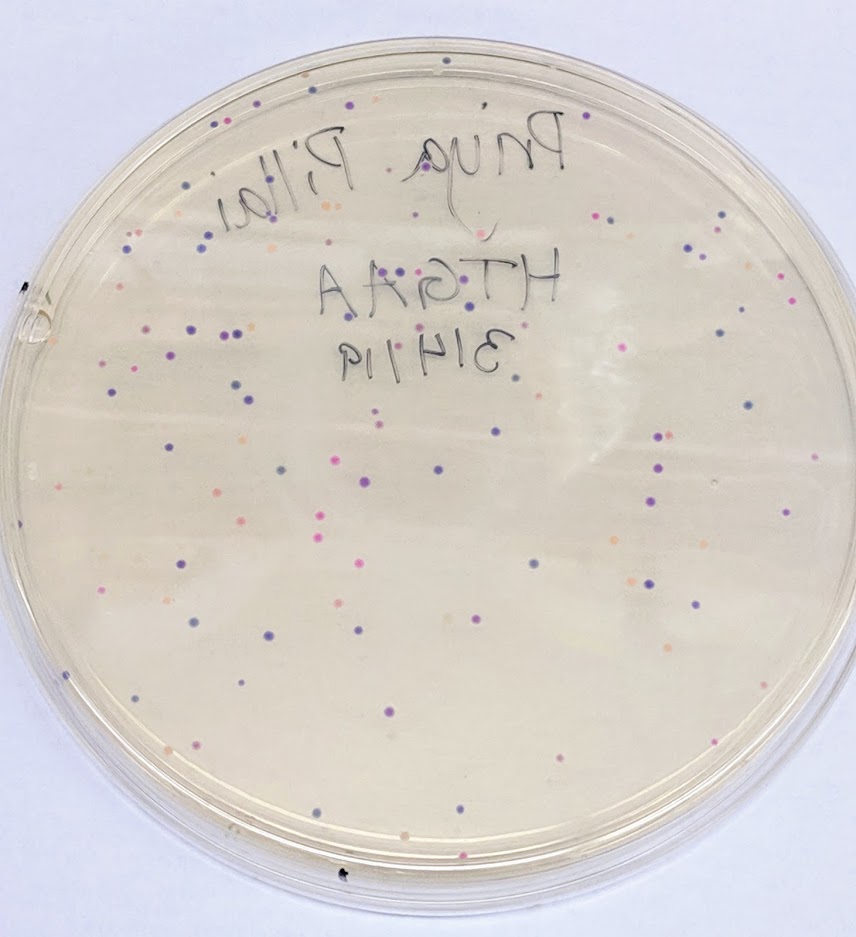
Once the final plasmid has been assembled with Gibson assembly, we heat shock E. coli. Heat shock causes the bacterial membrane to temporarily form pores that the plasmid can enter through. We expect the bacteria to only uptake one plasmid. However, this may not be a reasonable assumption as 10-beta cells are particularly well designed for large plasmids; it is possible a number of the E. coli took in 2 plasmids. Often, this is not an issue, as when the cell divides, it can split up the two plasmids, or one plasmid can outcompete the other. With the pUC19 backbone however, the ori is strong enough that the cells may be able to retain 2 different plasmids. We likely have a number of double transfectants.
If we were curious about the amount of mUAV that remained intact throughout the previous steps and was inserted into the bacteria, we could plate them on chloramphenicol plates, as the mUAV plasmids contain resistance to that antibiotic. To check if any colonies were double transfectants containing both the mUAV and modified pUC19 plasmids, we could replica plate the colonies onto another plate that also contained ampicillin. Any colonies that still survived on that plate would be the ones that also contained ampicillin.
