Week 6: Protein Design
In silico protein explorations
Protein analysis
Where did amino acids come from before enzymes that make them, and before life started? No one knows for sure yet, just as we don't know exactly how life began, but there's a very comprehensive review on the various hypotheses and evidence addressing this question that I will draw heavily upon.
- One theory posits that the earliest self-replicating system (RNA or something simpler) selected for amino acids from the pool of organic compounds that formed abiotically from the conditions and elements of prebiotic earth.
- Certain amino acids have been discovered in material identified to be extraterrestrial in origin; millenia of heat and radiation could have changed some to fill out the roster of amino acids we have today.
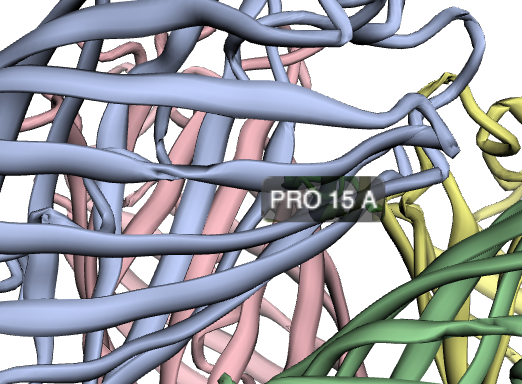
An exploration of the azoreductase 1 enzyme (cleaves azo bonds found in many dyes, causing the dye to decolorize) from the bacteria Pseudomonas putida, called azoR1.
A good place to start finding information about proteins (such as whether they have protein structures available) is UniProt.- The characterized protein is 203 amino acids long, with the most common amino acid being Alanine, comprising nearly 19% of the protein.
- A large of the protein structure is classified as "flavodoxin-like", which Pfam classifies as the "flavodoxin_2" protein family.
- There are more than a hundred protein homologs, one of which is a similar protein in the same organism, others which are the same protein in a different organism, and still others that are similar proteins in different organisms. In addition to using BLASTp to find homologous protein sequences, and using Clustal Omega to align the sequences to see where the conserved domains are and where differences exist, one can also tell Clustal Omega to produce a phylogeny tree from the aligned sequences as a prediction of how the proteins relate evolutionarily.
- The protein structure was solved by x-ray diffraction and deposited in the RCSB database in 2013. The quality of the protein varies depending on the metric; the resolution is 1.9 angstroms, which is about the diameter of an atom. RCSB classifies the protein as an oxidoreductase. The solved structure also includes two ligands: dodecaethylene glycol and a flavin mononucleotide (FMN) derivative.
I chose to visualize with NGL. To add display methods: main menu icon -> Representation -> select method. To color by secondary structure: menu icon -> colorscheme -> sstrc.
- There appear to be 12 alpha helices and 2 beta sheets.
- Color by hydrophobicity: colorScheme -> hydrophobicity; I determined that the default is that darker means less hydrophobic by checking specific residues. The protein is therefore mostly hydrophilic, which makes sense since it's active in water-soluble dye. Hydrophobicity seems to alternate by residue, and the white (hydrophobic) residues tend to be on the same side of a given helix.
- I found a few deep pockets that could be binding pockets; they all seem to have a hydrophobic residue inside, which makes sense as it would differentiate the binding pocket from the predominantly hydrophilic protein.
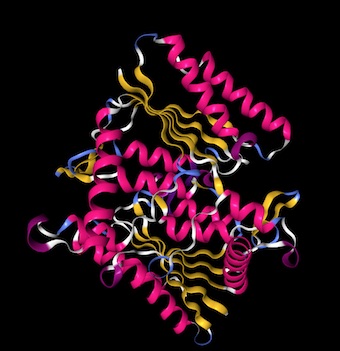
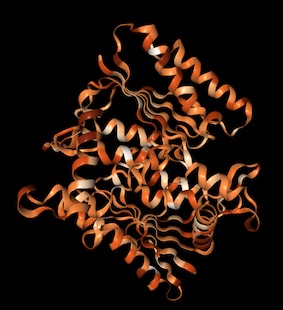
Protein structure prediction with Phyre 2
I used Phyre 2 to predict the structure of the amilCP fluorescent protein we worked with in Week 4. Phyre uses homology matching to known protein structures in its prediction algorithm. I also predicted the structure of a light blue mutant that we found on one student's plate, which turned out to have an additional non-chromophore mutation. I used PyMOL to visualize the structures, colored by secondary structure, highlighted the residues where the mutations occurred, and showed them as sticks (primer on selecting residues with commands in PyMOL).
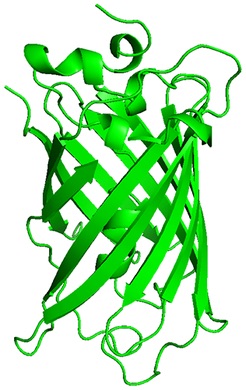
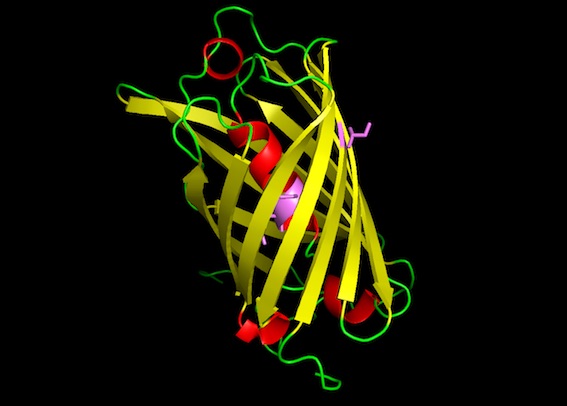
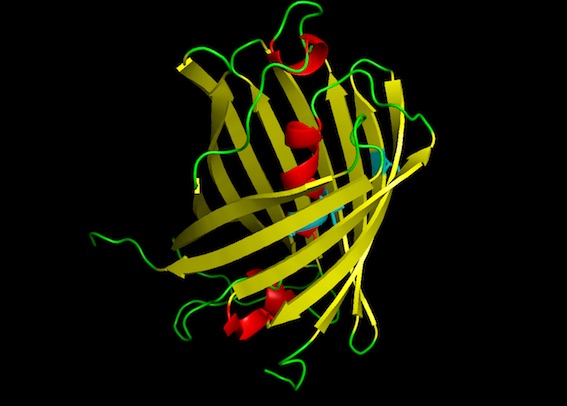
The predicted amilCP structure is very similar to GFP: beta-sheet barrel with an alpha-helix inside, and two residues in the middle of the alpha-helix being the chromophores. The mutant amilCP is not noticeably different.
Protein structure prediction with Robetta
Robetta is a protein structure web server that is built on Rosetta. I got slightly different results here than I did for Phyre. Notably, Robetta actually predicted a difference in the structure for our light blue mutant from the wild-type structure as a result of the non-chromophore mutation in the barrel (S15P): there is a twist at the nearby Tyr-13 residue in the light blue mutant amilCP structure that is not present in the wild-type. Noah referred me to literature that discusses how proline is an established, potent breaker of alpha-helical structures.
Wild-type amilCP
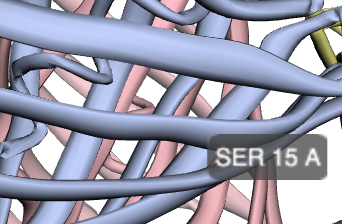
Light blue mutant amilCP