HTGAA: Next Generation Synthesis
- Joseph Jacobson (MIT)
Class Material
Noah Jakimo, Suryateja Jammalamadaka, Jessica Weber, Parnian Barekatain, Devora Najjar, and Berenice Estrada
Introduction
We will be changing the color-generating chromophore of the purple Acropora millepora chromoprotein (amilCP) to a variety of orange, pink, and blue mutants. These divergently-colored genetic variants of amilCP were described by Liljeruhm et al in 2018. Their strategy to identify where to mutate amilCP was inferred by sequence similarities to the chromophore region that allows for spectral engineering of the structurally-characterized and well-known green fluorescent protein (GFP), which is native to the jellyfish Aequorea victoria. First we will prepare for a Gibson assembly by using polymerase chain reaction (PCR) to produce two sets of amplicons as inserts and a restriction digest of the common cloning plasmid pUC19 to produce a new backbone (i.e. origin of replication and drug resistance gene). As template, both reactions use the amilCP-encoding plasmid that was miniprepped from the Addgene mUAV sample (deposited by the Nakayama lab at the University of Edinburgh and related to their paper on Mobius Assembly via a Mobius Assembly Universal Acceptor Vector). One set of amplicons copy the region of the amilCP gene that precedes the chromophore, including the transcription promoter and translation ribosome-binding site (RBS). Another set amplicons copy the region that spans 24 basepairs before the chromophore to just beyond the gene's transcription terminators. The latter includes a diversified chromophore-coding segment dictated by mismatches in the PCR primers with respect to the mUAV DNA template. The amplicon sets both include one end that overlaps by 20-22 bases with distinct ends of the large backbone fragment from the pUC19 digest. Lastly, we will express our colorful variety of amilCP mutants in electrocompetent E coli cell.
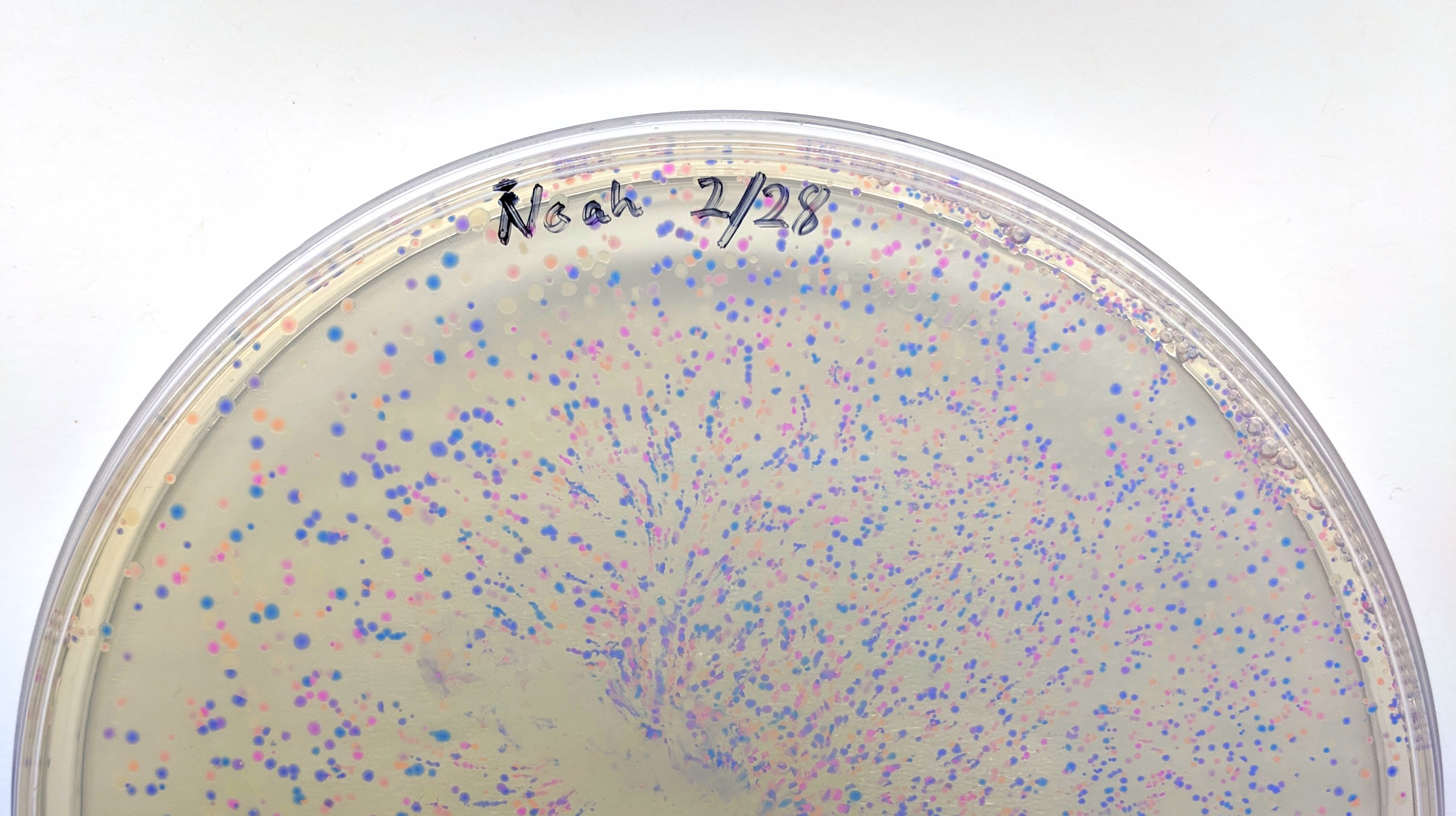
Primer Design
Design primers to amplify two sets of amplicons from mUAV plasmid. The amplicons sets must include one end that overlaps by 20-22 bases with distinct ends of the pUC19 backbone.
- Import Plasmid mUAV (addgene) and Plasmid pUC19 (addgene) into Benchling. Benchling tutorials
- Restriction digest pUC19 with PvuII and identify the backbone you want to use for your assembly. [Hint: You need a selection marker and origin of replication!]
- Identify the gene encoding for chromophore, RBS, promoter, and terminator in Plasmid mUAV.
-
In mUAV plasmid, if you change bases from 2309-2314 as shown below, you will get a wide variety of chromophore mutants with different colors. You design two sets of primers. One set amplifies the region that precedes the chromophore (before 2309) before these bases and the amplicon should consist of a part of the gene, RBS, and promoter. Another set amplicons copy the region that spans 24 basepairs before the chromophore
to just beyond the gene's transcription terminators. The latter includes a diversified chromophore-coding segment dictated by mismatches in the PCR primers with respect to the mUAV DNA template. The mismatches in primers can be designed using the following table.
Type Bases (2309-2314) Original TGTCAG mutant1 GTCAAC mutant2 TTTATG mutant3 GTCGGG mutant4 GTCCTG mutant5 GTCTCG mutant6 GCTTGT mutant7 TCAATG - The amplicon sets both include one end that overlaps by 20-22 bases with distinct ends of the large backbone fragment from the pUC19 digest you identified in the second step.
Experimental part
Remember to always initial and label your tubes so you can identify their contents and distinguish them from your classmates' material.
- Mini-prep mUAV plasmid from the overnight cultures.
-
Digest the pUC19 plasmid with the restriction enzyme PvuII to generate the linear blunt-ended backbone fragment.
-
Prepare the reaction in a PCR tube (add enzyme last and water first):
To Reaction Volume (50 uL Totral) DNase/RNase-Free Water 40 uL 2 ug pUC19 (1 ug/ul in green cap tubes) 2 uL 10x NEB Buffer CutSmart 5 uL NEB PvuII-HF 3 uL - Incubate reactions at 37C for at least 15 minutes in thermocycler.
-
Prepare the reaction in a PCR tube (add enzyme last and water first):
-
Amplify PCR fragments for the gene assembly of amilCP mutants.
-
Prepare the reaction in a PCR tube to produce the universal chromophore-preceding fragment:
To Reaction Volume (50 uL Totral) DNase/RNase-Free Water 20-X uL 200 ng miniprepped amilCP X uL Forward primer with tail that overlaps with cut pUC19 (10 uM in yellow cap labeled "TailVF Fwd") 2.5 uL Reverse primer preceding the chromophore (10 uM in yellow cap labeled "CPUni Rev") 2.5 uL 2x Phusion High-Fidelity PCR Master Mix 25 uL -
Prepare the reaction in a PCR tube to produce the chromophore mutant library fragments:
To Reaction Volume (50 uL Totral) DNase/RNase-Free Water 20-Y uL 200 ng miniprepped amilCP Y uL Reverse primer with tail that overlaps with cut pUC19 (10 uM in purple cap labeled "TailVR Rev") 2.5 uL Forward primer defining the chromophore library and overlaps with the universal reverse primer (10 uM in purple cap labeled "CPLib Fwd") 2.5 uL 2x Phusion High-Fidelity PCR Master Mix 25 uL -
Thermocycle reactions in a PCR program with with following program.
- Initial heat denature DNA at 98C for 30 seconds
-
Repeat the following for 35 cycles:
- Denature DNA at 98C for 10 seconds
- Anneal primers at 60C for 20 seconds
- Extend from primers with DNA polymerase primers at 72C for 1 minute. (Account for 1kb/30sec processivity)
- Final extension at 72C for 5 minutes.
- Hold at 4C until samples are retrieved.
-
Prepare the reaction in a PCR tube to produce the universal chromophore-preceding fragment:
-
Purify the DNA products from your reactions using the Zymo DNA Clean & Concentrator™-25 Kit. The following protocol is copied from Zymo Research and based on silica adsorption.
- For each reaction: In a 1.5 ml microcentrifuge tube, add 5x volumes of DNA Binding Buffer to each volume of reaction. Mix briefly by vortexing.
- Transfer each mixture into a separate Zymo-Spin™ Column in a Collection Tube.
- Centrifuge for 30 seconds at 13,000 rpm (~17,900 x g). Discard the flow-through.
- Add 200 µl DNA Wash Buffer to the column. Centrifuge at for 30 seconds at 13,000 rpm (~17,900 x g). Repeat the wash step.
- Transfer the column to a new 1.5 ml microcentrifuge tube and add 25 µl of DNase/RNase-free water directly to the column matrix. Let sit at room temperature for one minute.
- Centrifuge for 30 seconds at 13,000 rpm (~17,900 x g) to elute.
-
Measure the concentration of your purified DNA using the Nanodrop spectrophotometer.
- Clean stage with Kimwipe soaked with DI water.
- Wipe stage with dry Kimwipe.
- Add 2 uL DI water on stage and make blank measurement.
- Wipe stage with dry Kimwipe.
- For each DNA sample, add 2 uL and measure and record the concentration (ng/uL). Wipe stage dry with Kimwipe between measurements.
- To finish, clean stage with Kimwipe soaked with DI water and then wipe dry.
-
Setup your Gibson Assembly reaction in PCR tubes.
- In a 10 ul total volume, mix 100 ng of cut vector (PvuII digest) with a 2-fold excess of gene fragment (PCR reactions) in DNase/RNase-free water. Molar ratios can be approximated by the lengths of the DNA products. Note, the large fragment of the digested pUC19 is 2,364 bp. The gene fragment amplicons are 353 bp and 777 bp. (( [ng of vector] x [kb size of insert] ) / ( kb size of vector )) x ( insert:vector ratio) = ng of insert required
- Combine with 10 ul 2x Gibson Master Mix.
- Incubate in the thermocycler at 50C for at least 15 minutes.
-
Electroporate assemblies into electrocompetent E coli (stored across the hall in the -80C).
- Move Gibson assembly into a cold block on ice. Keep electroporation cuvettes, 10% Glycerol and microcentrifuge tubes (you can also use a PCR strip) on ice. Pre warm LB+ampicillin plates at 37C.
- Thaw electrocompetent cells on ice for 15-20 minutes.
- Aliquot 75 ul of 10% Glycerol followed by 25ul of competent cells into microcentrifuge tubes kept in a cold block on ice.
- Add 5 µl of the assembled product to the competent cells. Mix gently by pipetting up and down or by flicking the tube 4–5 times. Do not vortex.
- Transfer the mix to electroporation cuvettes without bubbles. Tap them gently to make sure the mix settles at the bottom part of the cuvette.
- Electroporate at 1700-2000V with 5ms time constant.
- Immediately transfer 900ul of SOC into the cuvettes. Pipette back and forth.
- Transfer it into a new 1.5 ml microcentrifuge tube. Incubate them for one hour at 37C.
- Spread 100-250 µl of the cells onto the ampicillin selection plates.
- Incubate overnight at 37C in the warm room.
Pro Challenge
Individually test the PCR performance of a set of shifted forward primers that change the chromophore-encoding sequence and have the template-mismatched positions closer towards the 3' end of the primer. Evaluate by gel electrophoresis.
Discussion Questions
- What are the Gibson overlap sequences in our DNA assembly design? How is an overlap created between the cut pUC19 and the PCR amplified parts of the amilCP gene?
- What is the melting temperature for each overlap sequence and how do they compare to the incubation temperature for the Gibson assembly reaction? See http://biotools.nubic.northwestern.edu/OligoCalc.html
- What is the purpose of each enzyme in the Gibson assembly mix?
Bonus Questions
- Our 50 ul PCR reactions included 0.5 uM of both forward and reverse primer and 200 ng of template DNA in the form of a 2,924 bp plasmid. Assuming perfect doubling of DNA copies with each PCR cycle, how many cycles does it take to use all primers? Recall, primers are extended to form copied products. See https://nebiocalculator.neb.com
- After the electroporation step in our experiment, how can we select for bacteria that contain only the original mUAV plasmid?
- Read the following post and its references: https://bitesizebio.com/2267/plasmid-retention/ Comment on the likelihood of a colony containing two plasmids from our electroporation into NEB 10-beta cells.
Useful Resources
- Demo videos for essential synbio skills: Synthetic Biology One
Related Readings & References
- A synthetic oscillatory network of transcriptional regulators
- Photoelectrochemical synthesis of DNA microarrays
- Accurate multiplex gene synthesis from programmable DNA microchips
- Maskless Fabrication of light-directed oligonucleotide microarrays using a digital micromirror array
- The crystal structure of DNA mismatch repair protein MutS binding to a G·T mismatch
- Protein-mediated error correction for de novo DNA synthesis
- Enzymatic assembly of DNA molecules up to several hundred kilobases
- Scalable gene synthesis by selective amplification of DNA pools from high-fidelity microchips
- Multiplexed gene synthesis in emulsions for exploring protein functional landscapes
- Parallel gene synthesis in a microfluidic device
- De novo DNA synthesis using polymerase-nucleotide conjugates
- Genomically Recoded Organisms Expand Biological Functions
- In vivo cleavage rules and target repertoire of RNase III in Escherichia coli
- High-level semi-synthetic production of the potent antimalarial artemisinin
- Multiplex single-molecule interaction profiling of DNA-barcoded proteins