HTGAA: Protein Design
- Shuguang Zhang (MIT)
- Thras Karydis (DeepCure)
Class Material
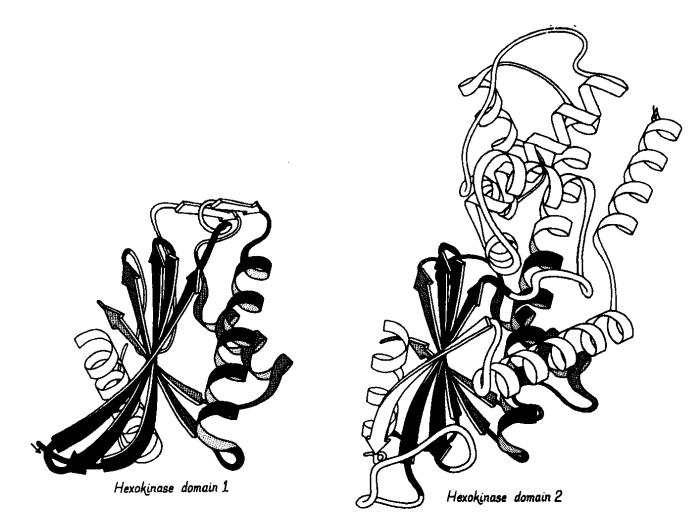
Jane S. Richardson's Anatomy and Taxonomy of Protein Structure (1981)
This week is all about proteins! The homework is divided into three parts. Part A is focused on protein analysis and protein informatics. In Part B, you will have a fun introduction to the challenging world of protein folding. Finally, in Part C, you will get run state of the art machine-learning models for protein design!
Part A: Protein Analysis
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins.
Exercises
-
Answer any of the following questions
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
- Why are there only 20 natural amino acids?
- Why most molecular helices are right handed?
- Where did amino acids come from before enzymes that make them, and before life started?
- What do digital databases and nucleosomes have in common?
-
Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions.
-
Briefly describe the protein you selected and why you selected it.
-
Identity the amino acid sequence of your protein.
-
How long is it? What is the most frequent amino acid?
-
How many protein sequence homologs are there for your protein?
Hint: Use the pBLAST tool to search for homologs and ClustalOmega to align and visualize them.
-
Does your protein belong to any protein family?
-
-
Identify the structure page of your protein in RCSB
- When was the structure solved? Is it a good quality structure?
- Are there any other molecules in the solved structure apart from protein?
- Does your protein belong to any structure classification family?
-
Open the structure of your protein in any 3D molecule visualization software
- Visualize the protein as "cartoon", "ribbon" and "ball and stick".
- Color the protein by secondary structure. Does it have more helices or sheets?
- Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
- Visualize the surface of the protein. Does it have any "holes" (aka binding pockets)?
-
Resources
Databases
- Uniprot: A comprehensive, high-quality and freely accessible resource of protein sequence and functional information. It is linked to almost every other database. Example Entry: G3ECR1
- RCSB: Collection of all publicly available biological macromolecular structures. Example Entry: 4CMP
- PFAM: A large collection of protein families, i.e. groups of proteins with similar sequence/function. Example Entry: PF02171
- SCOP: A large collection of structural protein families. Proteins are organized according to their structural and evolutionary relationships. Example Entry: 6PGDH C-terminal helical region-like
- ExPaSy: SIB Bioinformatics Resource Portal which provides access to scientific databases and software tools (i.e., resources) in different areas of life sciences including proteomics, genomics, phylogeny, systems biology, population genetics, transcriptomics etc.
3D Molecule Visualization software
-
PyMOL(https://pymol.org/edu/?q=educational): PyMOL is a user-sponsored molecular visualization system on an open-source foundation, maintained and distributed by Schrödinger.
- Practical PyMOL for Beginners
- Video Tutorials: Video 1 Video2 (and tons more… just search "PyMOL tutorial" in youtube).
- Cheat Sheet
- Advanced Cheat Sheet
-
Chimera: A highly extensible program for interactive visualization and analysis of molecular structures and related data, including density maps, supramolecular assemblies, sequence alignments, docking results, trajectories, and conformational ensembles.
- Chimera Tutorials
- Video Tutorials: Video 1 Video 2 (and tons more… just search "Chimera tutorial" in youtube).
-
VMD: A molecular visualization program for displaying, animating, and analyzing large biomolecular systems using 3-D graphics and built-in scripting
- VMD Tutorials
- Video Tutorials: Video 1 Video 2 (and tons more… you know the drill)
-
NGLViewer: NGL Viewer is a collection of tools for web-based molecular graphics. WebGL is employed to display molecules like proteins and DNA/RNA with a variety of representations.
Sequence alignment and homology
-
BLAST: BLAST finds regions of similarity between biological sequences. The program compares nucleotide or protein sequences (pBLAST) to sequence databases and calculates the statistical significance.
-
Clustal Omega: A new multiple sequence alignment program that uses seeded guide trees and HMM profile-profile techniques to generate alignments between three or more sequences.
-
The BLOSUM matrices are used during alignment to check how similar are amino acids to each other. Here is the BLOSUM62 matrix, most commonly used if no a-priori information is available for the evolutionary relationship of the protein sequences.
Part B: How to (almost) Fold (almost) Anything
In this part you will be folding protein sequences into 3D structures. The goal is to get an understanding on how computational protein modeling works as well as to see first hand the great computing power needed for molecular simulations in biology.
Folding Online (Robetta)
First, we will use an online Rosetta engine (Robetta) to get the feel of protein folding. Since folding is a computationally intense task, we will choose a protein with less than 100 amino-acids long, so we should get the result back in 2-3 days.
- Use RCSB to find a protein with less than 100aa. (Hint: try looking for PDB statistics...)
- Download the pdb file of your choice. And copy it's sequence (can be done in RCSB website, or alternatively in PyMOL).
- Go to Robetta. Register an account.
- Submit your sequence for folding.
- Get the results back in a couple of days.
- Compare to the known protein structure using PyMOL. Notice that while both proteins will have similar structures, they aren't necessarily aligned - you might have to move and rotate them first. Here is a short video tutorial on aligning structures with PyMOL. Post your results.
Here is how Robetta results look like: https://robetta.bakerlab.org/results.php
Pro Challenge: Folding Offline (PyRosetta)
By following these instructions, you can install Roestta on your machine and play around with folding proteins at quicker speeds compared to Robetta.
- Install PyRosetta by following the instructions below
- Git clone or download the following GitHub repository: https://github.com/thrakar9/protein_folding_workshop.
-
Folding a small (30 aa) peptide.
a. Open the "Protein Folding with Pyrosetta" Jupyter notebook. Execute interactively the code in the notebook and answer the questions therein. When you are done, save the notebook (with the answers and all outputs) to an HTML file, and link it to your class page.
b. Pick the lowest energy model and structurally (visually) compare it to the native. How close is it to the native? If its different, what parts did the computer program get wrong? Note: To compare the structures you have first to align them to the native. You can do that very easily in PyMOL. Here is a short video tutorial on aligning structures with PyMOL
c. Pick the lowest RMSD model and structurally compare it to the native. How close is it to the native? If its different than the lowest energy model, how is it different? Remember that in a blind case, we will not have the benefit of an RMSD column.
-
Fold your own sequence! In question 1 we used the sequence from a human protein as input to the folding algorithm. Yet, in principle, you can give any arbitrary sequence of amino acids as an input.
a. Use any process to create a sequence of 30-50 amino acids, and predict it's 3D structure using the notebook from Q1. You can try to run the script with multiple parameter combinations and compare the results. Log the parameters that had the best outcome.
b. Compare the resulting structures of 2(a) with those from question 1. Do the structures in both cases look protein-like ? If not, can you think of an explanation?
c. Try folding multiple sequences to come up with the most protein-looking structure!
For inspiration, last year Belen created and folded a protein that contains all of the names of the HTGAA.20 cohort! What can you come up with?
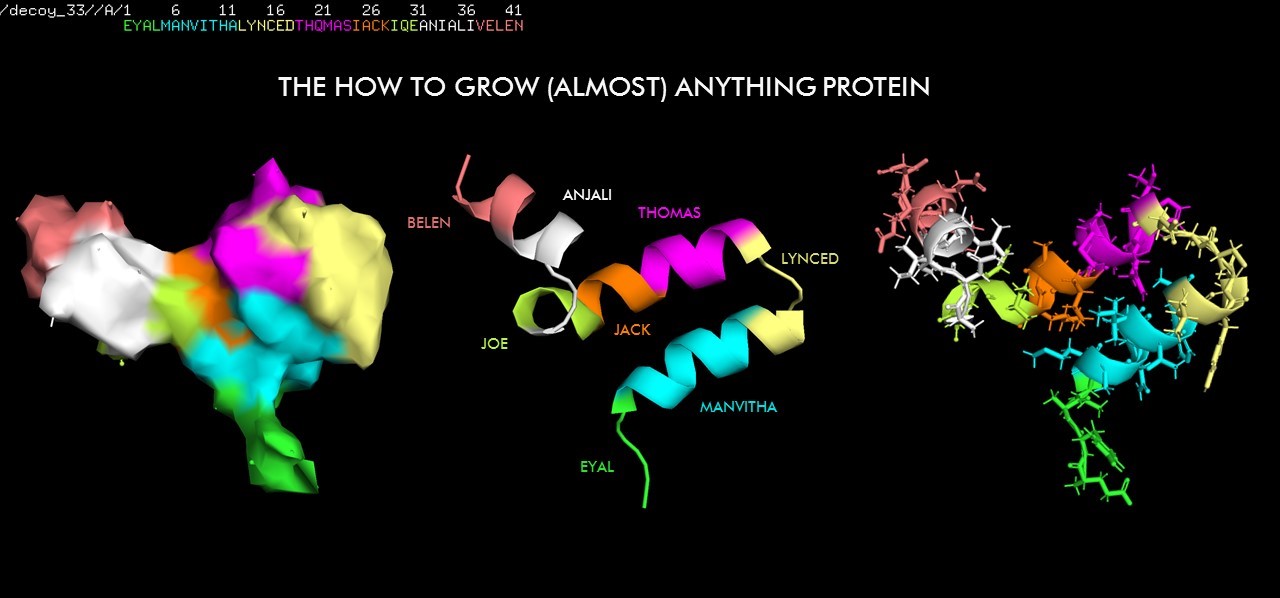
© Belen Vicente Blazquez 2020
3D Print Your Protein!
In this part, you can choose any protein structure (rather it's a solved one from RCSB, or one that you folded yourself!) and convert the structure file to an .stl file for 3D printing. We will be printing your protein at MIT using Formlabs and you can pick it up from the MIT Media Lab.
The details of the protocol can be found here: https://www.ascb.org/careers/3d-print-your-favorite-protein
Resources
Setting up PyRosetta
-
Download and install Anaconda.
- Visit https://www.anaconda.com/distribution/
- Select the Python 3.7 version and follow the instructions to install
-
Create a Python 3.6.8 virtual environment with conda
conda create -n protein_design python=3.6.8- Verify you have the correct Python version by activating the environment `conda activate protein_design` and executing the command
python. You should an output similar to this:
Python 3.6.8 | Anaconda, Inc. | (default, Dec 29 2018, 19:04:46)
[GCC 4.2.1 Compatible Clang 4.0.1 (tags/RELEASE_401/final)] on darwin
Type "help", "copyright", "credits" or "license" for more information.
Download and install PyRosetta
-
Select the Python 3.6 version for your system
-
Download. Username:
levinthaland PasswordparadoxNote: This combination of username/password is only for academic use.
-
Activate the virtual environment we created above:
conda activate protein_design -
Extract and install PyRosetta to the environment.
tar -vjxf PyRosetta-<version>.tar.bz2
cd setup && python3.6 setup.py install
- Verify the installation in Python.
python3.6 -c "import pyrosetta; pyrosetta.init()"
Working with Jupyter notebooks
Jupyter Notebooks are simply amazing. If you haven't used them before, today is your lucky day. Some resources:
- Video Tutorial
- You can save your notebook as an HTML file (
File->Download as->HTML)
Protein folding (structure prediction) webservers
Part C: Protein Design by Machine Learning
Finally, we would like to design proteins, or optimize existing proteins to have better (or worse) properties and abilities. Over the last few years, machine learning based approaches have revolutionized the protein folding and design field. In this part, we are going to follow along a Jupyter Notebook based on a paper by Facebook AI Research: Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences" (Rives et al., 2020)
Embeddings are a concept in machine learning that started in Natural Language processing and is now used to understand the "language" of protein sequences. Unsupervised ML model is a model that only gets data (X) without any labels, and tries to make sense of the patterns within the data. Embeddings are the product of an unsupervised ML model that tries to transform raw data (e.g. amino acid sequence) into a space while retaining useful properties (e.g. proteins with different biochemical properties will be further apart in that space). What's very useful in embeddings that we can use them for unseen data (e.g. new protein sequence) and get new insights. Another thing we can do is to "travel" within a space - given a protein's embedding, can we make it shine brighter? more hydrophobic? heat-resistant?
In this notebook, we are going to use pre-trained protein embeddings by FAIR to train a downstream model that predicts the effect of missense mutations on the sequence of the protein Beta-lactamase. The original data is from the Envision paper (Gray, et al. 2018)
- Open the Google Colab Notebook
- Click "Copy to Drive" to make a copy on your own Google Drive/Colab
- Follow along and run the notebook. While most of it doesn't require any changes, try to understand what each cells does and feel free to play around and break it
- Towards the end, you can choose one of three regressors (feel free to try others). Report in your homework which one you chose and what Spearman correlation score did you get
- What does this all mean? Why is this useful? Last, try to think and design an experiment where machine learning induced protein design can make an impact. Choose a protein of your choice from any animal, plant, fungi, bacteria or virus. Which properties of this protein could you enhance (or diminish) using in-silico design. Can you also think of ways to generate data in the wet lab to be digested by machine learning models (similarly to Rives 2020 and Bryant et al 2020).