HTGAA: Next Generation Synthesis
Class Material:
- Class Slides
- Class Video
- Recitation #1 Video
- Recitation #2 Video
Introduction
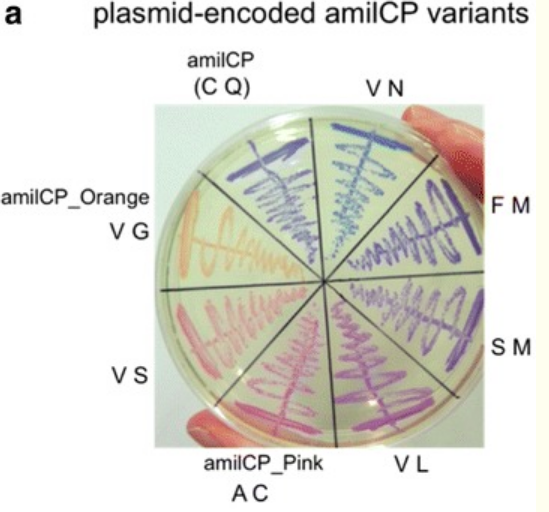
We will be changing the color-generating chromophore of the purple Acropora millepora chromoprotein (amilCP) to a variety of orange, pink, and blue mutants. These divergently-colored genetic variants of amilCP were described by Liljeruhm et al in 2018. Their strategy to identify where to mutate amilCP was inferred by sequence similarities to the chromophore region that allows for spectral engineering of the structurally-characterized and well-known green fluorescent protein (GFP), which is native to the jellyfish Aequorea victoria. First, we will prepare for a Gibson assembly by using polymerase chain reaction (PCR) to produce two sets of amplicons as inserts and a restriction digest of the common cloning plasmid pUC19 to produce a new backbone (i.e. origin of replication and drug resistance gene). As a template, both reactions use the amilCP-encoding plasmid that was miniprepped from the Addgene mUAV sample (deposited by the Nakayama lab at the University of Edinburgh and related to their paper on Mobius Assembly via a Mobius Assembly Universal Acceptor Vector). One set of amplicons copy the region of the amilCP gene that precedes the chromophore, including the transcription promoter and translation ribosome-binding site (RBS). Another set of amplicons copy the region that spans 24 basepairs before the chromophore to just beyond the gene's transcription terminators. The latter includes a diversified chromophore-coding segment dictated by mismatches in the PCR primers with respect to the mUAV DNA template. The amplicon sets both include one end that overlaps by 20-22 bases with distinct ends of the large backbone fragment from the pUC19 digest. Lastly, we will express our colorful variety of amilCP mutants in electrocompetent E coli cell.
 © Noah Jakimo 2019
© Noah Jakimo 2019
Part A: Primer Design and Fragment Assembly
In this part, we will prepare and order the primers that will generate a library of mutated amilCP expressing E. coli cells. We will use Gibson Assembly to insert our mutated gene into a plasmid, which in turn will be transformed into electrocompetent E. coli cells. First, let's understand what is Gibson Assembly and how to design primers for it.
Gibson Assembly
Video #1:
Video #2:
Excellent explanation (up until 8:00, the last part is less relevant for us):
Your Mission, Should You Choose To Accept It
Design primers to amplify two sets of amplicons from mUAV plasmid. The amplicons sets must include one end that overlaps by 20-22 bases with distinct ends of the pUC19 backbone.
- Import the pUC19 plasmid sequence from Addgene into Benchling
- Restriction digest pUC19 with PvuII and identify the backbone you want to use for your assembly. [Hint: You need a selection marker and origin of replication!]
- Import the mUAV plasmid sequence into Benchling, by going to Import DNA Sequences > Search External Databases and input the GenBank identifier MG252981.1
- Interestingly, we will be actually be using a Twist Gene Fragment as the source DNA. For our 1kb fragment, both the price and delivery times are better compared to plasmids, and importantly, require less TA lab work (no need to miniprep). To examine which fragment we ordered, use this link: https://benchling.com/s/seq-uivVVxZrv3WxMWNhyTJu
- Identify the amilCP gene, RBS, promoter, and terminators in Plasmid mUAV.
- As described in Liljeruhm et al, the amilCP gene contains a chromophore (CP) region that can be mutated to express different colors. The mutation region is: cagTGTCAGtac
- Identify the CP mutation sequence (TGTCAG) in the gene and annotate it.
- Use a codon table and convert the following figure to a table of colors of DNA sequences
Notice that there's more than one right answer! One amino acid sequence can be coded by multiple DNA sequences!
- We will split create two fragments out of the mUAV plasmid. One fragment will contain the RBS, promoter and first part of amilCP gene right up until the CP mutation region. The second fragment will include the CP mutation all the until the terminators.
- To generate these two fragments, we will design four primers (two forward, two reverse). Each of the primer sequences should follow the primer design guidelines to increase your chances of success in the experiment. A few guidelines as taken from here
- Primer Length: It is generally accepted that the optimal length of PCR primers is 18-22 bp. This length is long enough for adequate specificity and short enough for primers to bind easily to the template at the annealing temperature. However, in our case we will design long overhangs to prepare for Gibson Assembly, so the binding region of the primer should be 18-22 bp, followed by a 20-22 bp overhang.
- Melting Temperature (Tm): the temperature at which one half of the DNA duplex will dissociate to become single stranded and indicates the duplex stability. Primers with melting temperatures in the range of 52-58C generally produce the best results. Primers with melting temperatures above 65C have a tendency for secondary annealing. Importantly, primers in the same set should have a similar Tm (5C) between each other!
- GC clamp: The presence of G or C bases within the last five bases from the 3' end of primers (GC clamp) helps promote specific binding at the 3' end due to the stronger bonding of G and C bases. More than 3 G's or C's should be avoided in the last 5 bases at the 3' end of the primer.
- GC content: the number of G's and C's in the primer as a percentage of the total bases should be 40-60%.
- Primer Secondary Structures (a.k.a primer dimer): Presence of the primer secondary structures produced by intermolecular or intramolecular interactions can lead to poor or no yield of the product. They adversely affect primer template annealing and thus the amplification. They greatly reduce the availability of primers to the reaction.
- Benchling allows us to check for secondary structure by selecting part of the sequence > Create Primer > Check Secondary Structure
- Another great online software is NUPack.
- It can be quite hard to design primers with no secondary structures. A rule of thumb is to keep the Gibbs free energy of each structure at above -10kcal. For your task, just report what are the secondary structures did you get for your primer pairs.
- For more info and a video: https://www.addgene.org/protocols/primer-design/
- In your report, elaborate how you chose your primers and according to these design guidelines. Notice that we can't always make every primer perfect, but the more guidelines you follow the higher your chances of success.
Primers
- Outer Forward Primer:
- mUAV: identify an 18-22bp region just before the promoter/RBS. Note that you can see your GC content and Tm on the bottom. Right-click and Create Primer (Forward) to examine the design parameters. Copy the sequence to a text editor.
- pUC19: identify the ~20bp region just after the PvuII cut site.
- Combine these sequences to get your Outer Forward Primer
- Make sure your 5 to 3 orientation is right, this is very confusing!
- Outer Reverse Primer:
- mUAV: identify an 18-22bp region just after the terminators. Right-click and Create Primer (Reverse) to examine the design parameters. Copy the sequence to a text editor.
- pUC10: identify the ~20bp region just after the PvuII cut site.
- Combine these sequences to get your Outer Reverse Primer
- Make sure your 5 to 3 orientation is right, this is very confusing!
- Inner Reverse Primer:
- mUAV: identify the chromophore (CP) mutation region
- Select a 18-24bp region just before the CP mutation region. Right-click and Create Primer (Reverse).
- Copy the sequence to a text editor to get your Inner Reverse Primer
- Inner Forward Primer (Mutations):
- mUAV: identify the chromophore (CP) mutation region
- Select a 18-24bp region before and after the CP mutation region, as well the the CP mutation region (total length: 18-24 + 6 + 18-24 = 42-54bp!)
- This will be our PCR primer + overhang for Gibson Assembly.
- Copy this sequence to a text editor. Now, use the table you made above to choose which color variant you wish to express.
- You can have multiple colors together! each E.coli cell will only get one plasmid and express it, but we will have many cells and a variety of colors. You can also order different mixes. Go wild.
- To make a mutation library, you have to options:
- Prepare a bunch of primers, that will be synthesized seperately and you will later mix them together using the robot.
- Use degenerate bases, where a single letter (e.g. H) means it could be a number of different bases (e.g. A/C/T).
- For example, if we synthesize the sequence HTGAA, we will get a mixture of: ATGAA, CTGAA, TGTAA in a single tube.
- Your set of sequences is your Inner Forward Primer
This week's task is purely in-silico design. Next week we will use the same primers you designed and perform fragment assembly using the remote robot. We will send your designed primers for DNA synthesis. Please send all of your primers (simply as text files) to Eyal by 03/22 09:00, so we can order them and have the experimental setup ready by next Wednesday. Your number of primers can be between four to ten per student. Notice that if you use degenrate bases, you can have more than "one" primer sequence that still counts as a single order. For example, ordering HTGAA will give you a single tube that contains three primers: ATGAA, CTGAA, TGTAA

Part B: Remote Cloning
Final Projects and Twist Genes
In this exercise, we are extracting a specific gene (amilCP) from a plasmid and mutating it using PCR. As we talked about in class, Next Generation DNA synthesis is changing the way we think about bio-design. Twist Bioscience, as part of its gracious support to HTGAA, offers us a special budget for ordering gene fragments and clonal genes. These could be extremely handy for your final projects. Essentially, you could choose any gene you fancy and submit it to Twist. When ordering clonal genes, they already take care of the work of inserting it into a vector (plasmid). Meaning, you can just choose a gene and choose an bacterial expression vector and you will get a bacteria expressing your synthetic gene (with no lab work!)
More details can be found here:
Bonus Content
There are other ways to assemble a fragment library into a plasmid. Another common technique, which we will not be using this week (but keep in mind for final projects!) is Golden Gate Assembly
Golden Gate Assembly
We used NUPack to make sure there are no DNA-DNA interactions between our primers. Sometimes, we are interested in designing DNA that will interact with itself or with other DNA molecules. We can create molecular switches that respond to a signal or temperature. See this video for a short and great explanation on hairpin structures and toehold switches (this can definitely be part of a final project idea!):
Useful Resources
- Demo videos for essential synbio skills: Synthetic Biology One