This week is all about proteins! The homework is divided into two parts. Part A is focused in protein analysis and protein informatics. Part B, is about coumputational protein folding.
Part A: Protein Analysis and Protein Informatics
- Answer any of the following questions:
- How many molecules of amino acids do you take with a piece of 500 grams of meat(on average an amino acid is ~100 Daltons)?
- Why are there only 20 natural amino acids?
- Why most molecular helices are right handed?
- Where did amino acids come from before enzymes that make them, and before life started?
- What do digital databases and nucleosomes have in common?
- Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions
- Briefly describe the protein you selected and why you selected it.
- Identity the amino acid sequence of your protein.
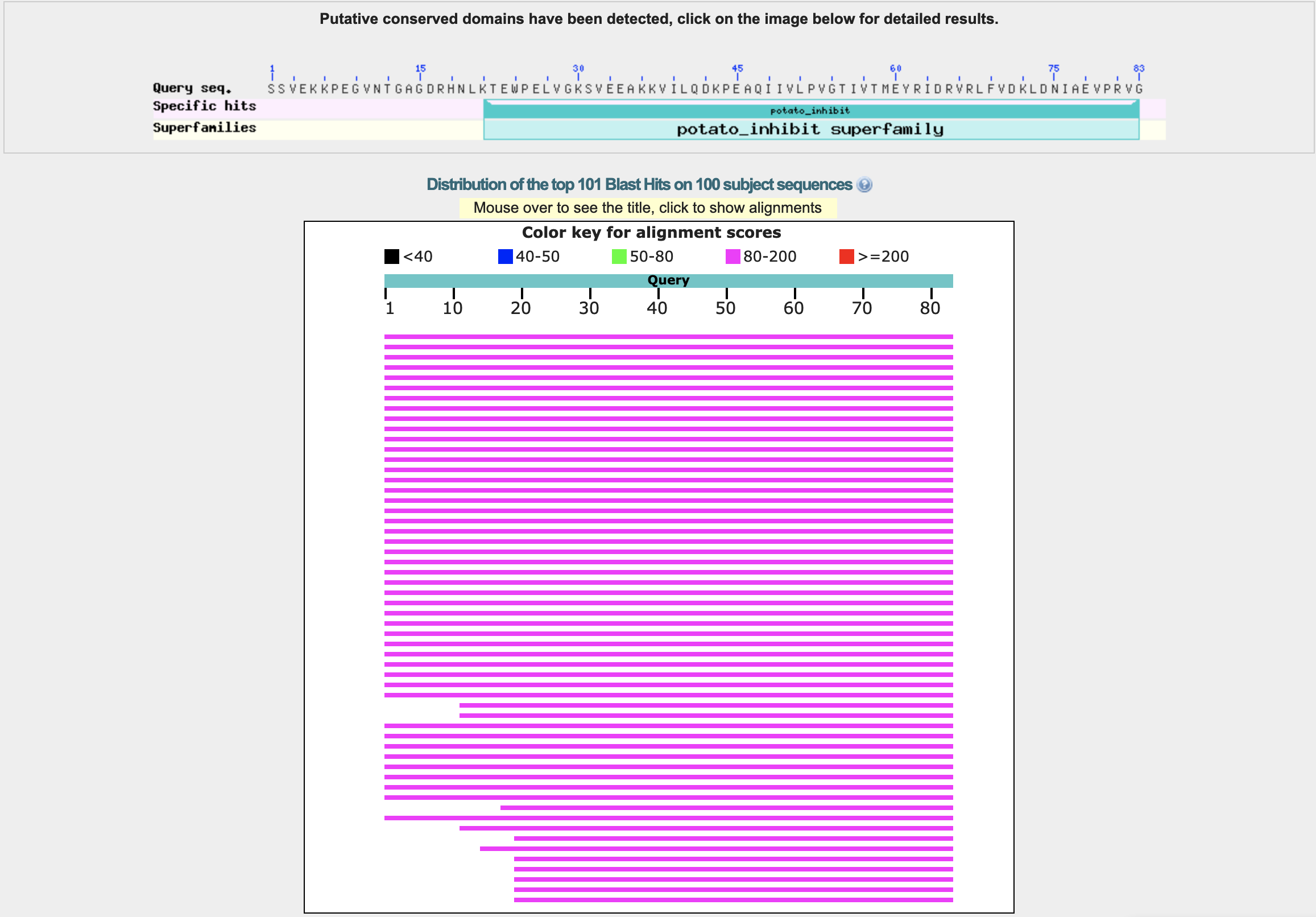
- How long is it? What is the most frequent amino acid? The length of the amino acid sequence is 83. The sequence is SSVEKKPEGVNTGAGDRHNLKTEWPELVGKSVEEAKKVILQDKPEAQIIVLPVGTIVTMEYRIDRVRLFVDKLDNIAEVPRVG. The most frequent amino acid is valine (V), which appears 12 times. The frequency distribution is the following: {’S’: 3, 'V': 12, 'E': 9, 'K': 8, 'P': 5, 'G': 6, 'N': 3, 'T': 4, 'A': 4, 'D': 5, 'R': 5, 'H': 1, 'L': 6, 'W': 1, 'I': 6, 'Q': 2, 'M': 1, 'Y': 1, 'F': 1}.
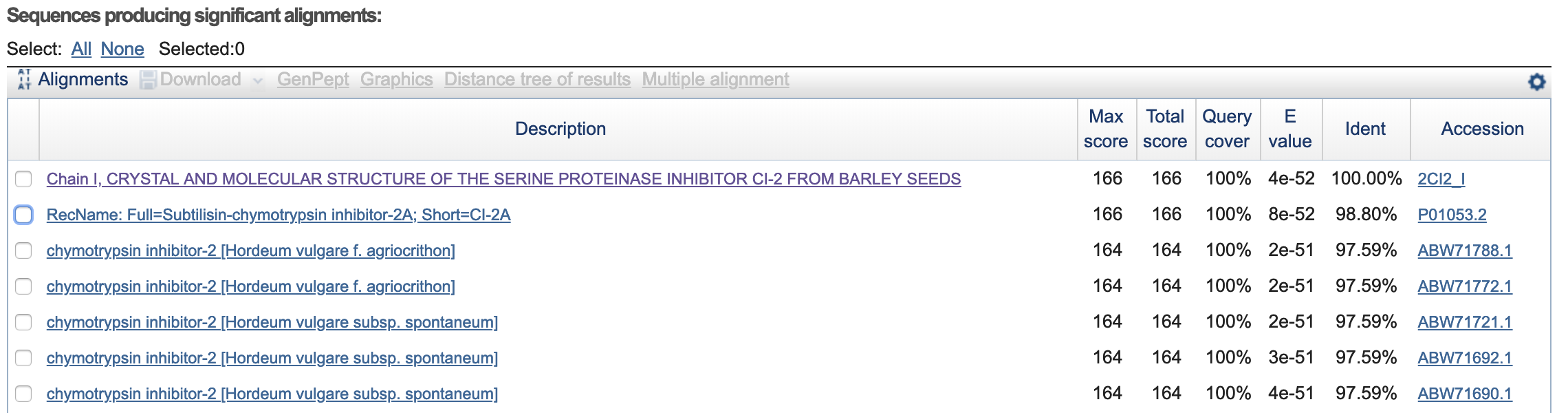
- How many protein sequence homologs are there for your protein?
- Does your protein belong to any protein family? Classification: Proteinase Inhibitor (Chymotrypsin). Organism(s): Hordeum vulgare. Its a member of the potato inhibitor 1 family. CI-2 family of serine protease inhibitors.
- Identify the structure page of your protein in RCSB
- When was the structure solved? Is it a good quality structure? 1988-09-05.
- Are there any other molecules in the solved structure apart from protein? No.
- Does your protein belong to any structure classification family? No.
- Open the structure of your protein in any 3D molecule visualization software
- Visualize the protein as "cartoon", "ribbon" and "ball and stick".
- Color the protein by secondary structure. Does it have more alpha helices or beta sheets?
- Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
- Visualize the surface of the protein. Does it have any "holes" (aka binding pockets)?
Assuming that the average molecular weight of each amino acid (M) is 100 Daltons or g/mol and mass (m) is 1g, leads to n = m/M = 0.01 mol of amino acids. Taking Avogadro’s constant 1 mol = ~6.022𝐸23 mol(-1) then, X = 0.01 mol * 6.022𝐸23 mol(-1) = 6.022𝐸21 amino acids. Considering that 100 g serving of red meat provides around 28g of protein, this approximates to 3.011E26 amino acid molecules.
The choice of why only 20 still remains a mistery. Francis Crick proposed in the 60s, that the choice of amino acids was arbitrary, suggesting that a different group of 20 would be just as good. Recent studies suggest that the 20 amino acid properties, such as hydrophobic vs polar, size, charge, etc. are evenly distributed through the chemical space. Other theories suggest that other amino acid structures could have been used. Another interesting question is how the amino acids are encoded in the nucleotide code. The smallest combination of four bases that could encode all 20 amino acids would be a triplet code. However, a triplet code produces 64 (43 = 64) possible combinations, or codons. Thus, a triplet code introduces the problem of being more than three times the number of codons than amino acids. There are few cordons that encode just one amino acid, such as the codon for tryptophan. In addition, some cordons encode start and stop information. So, one hypothesis could be that nature often resorts to redundancy for encoding information. More Info.

Some experimental and modelling observations suggests, folding energy for right handed in more favorable. Additional information here and here.

The protein I picked is the Crystal and molecular structure of the serine proteinase inhibitor CI-2 from barley seeds (2CI2). I picked this protein because of the interesting ring-like structural shape. More info on the protein here.
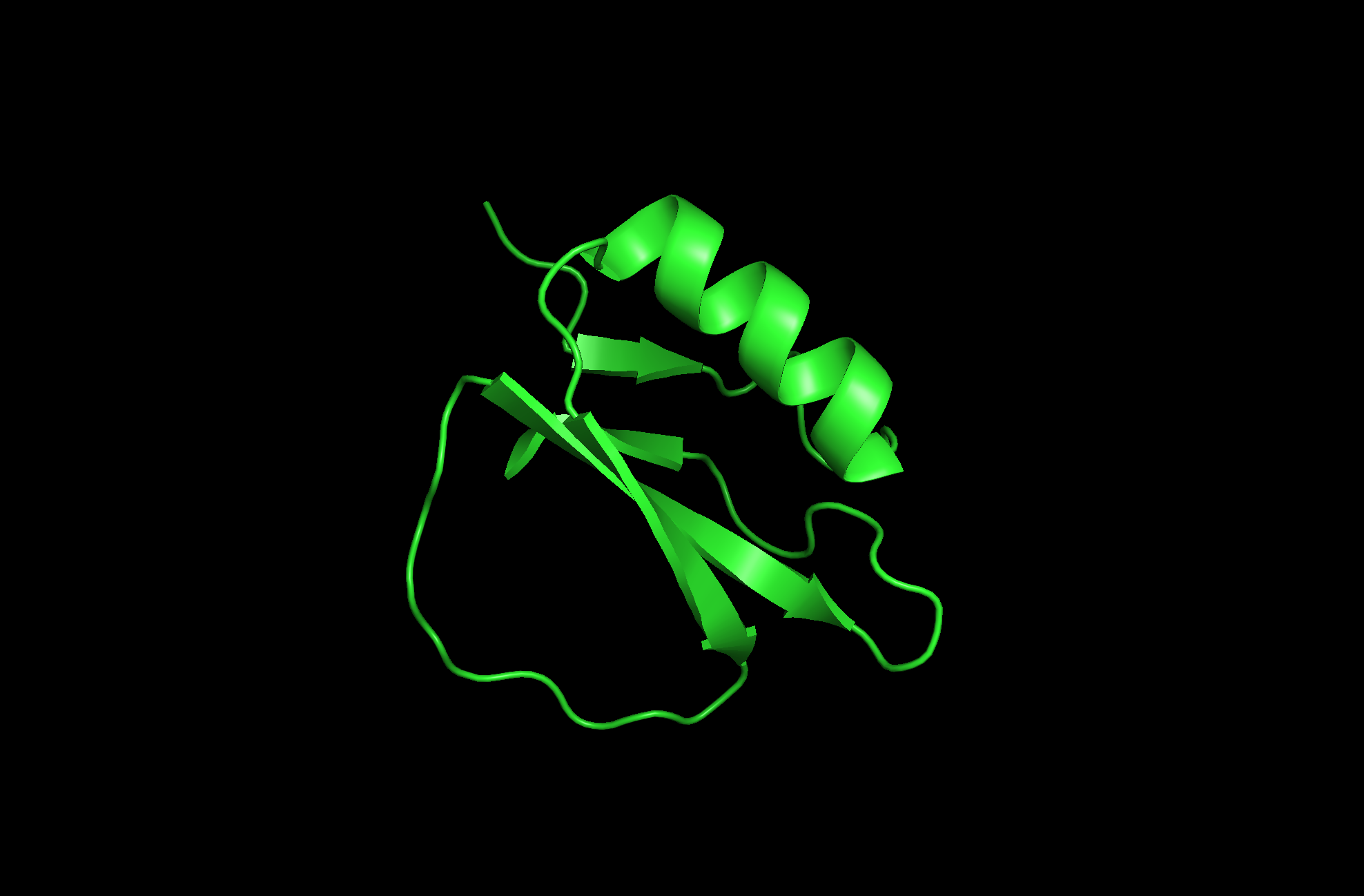
2CI2 Cartoon Visualization
2CI2 Lines Visualization

2CI2 Ribbon Visualization
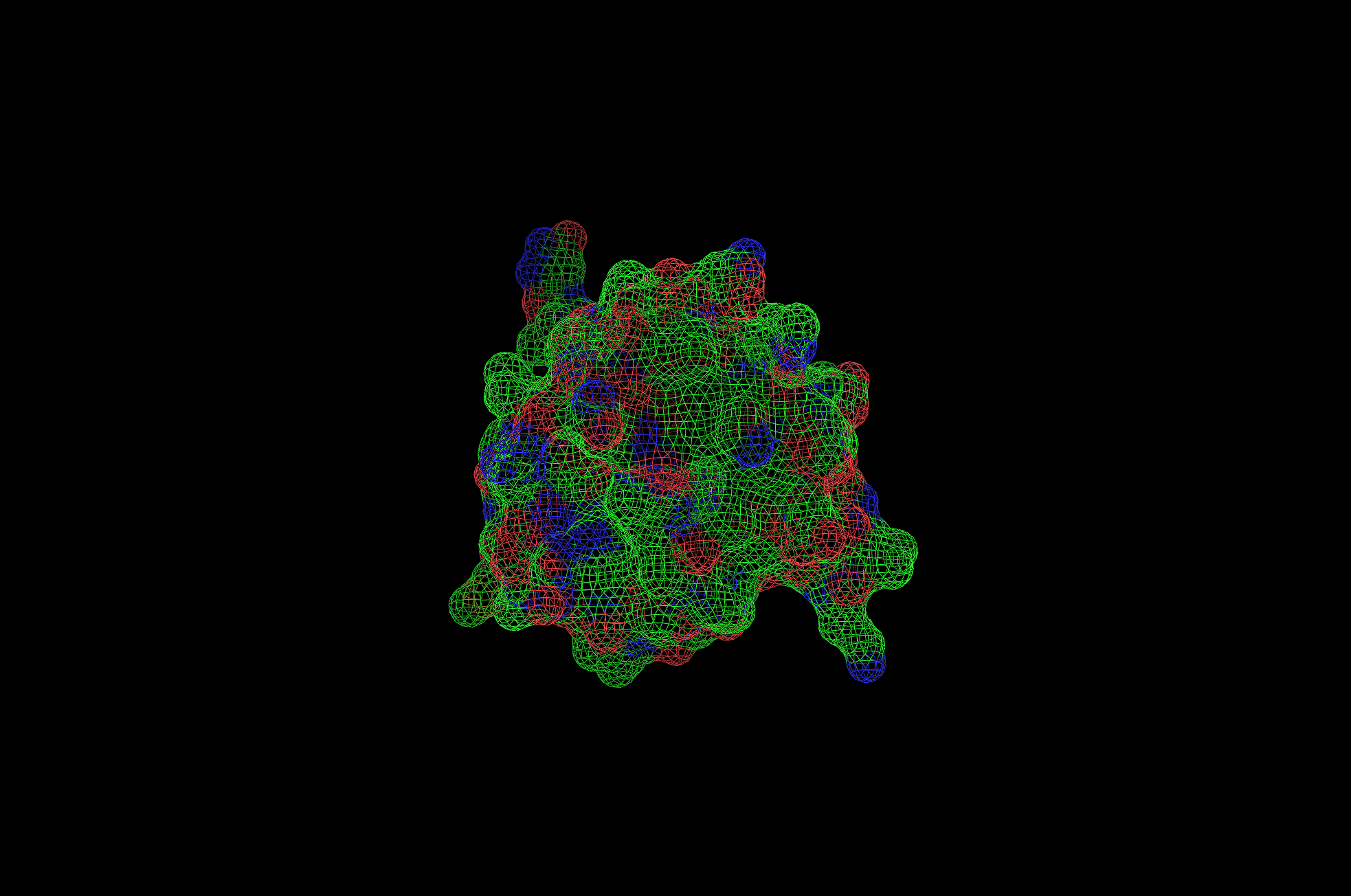
2CI2 Mesh Visualization
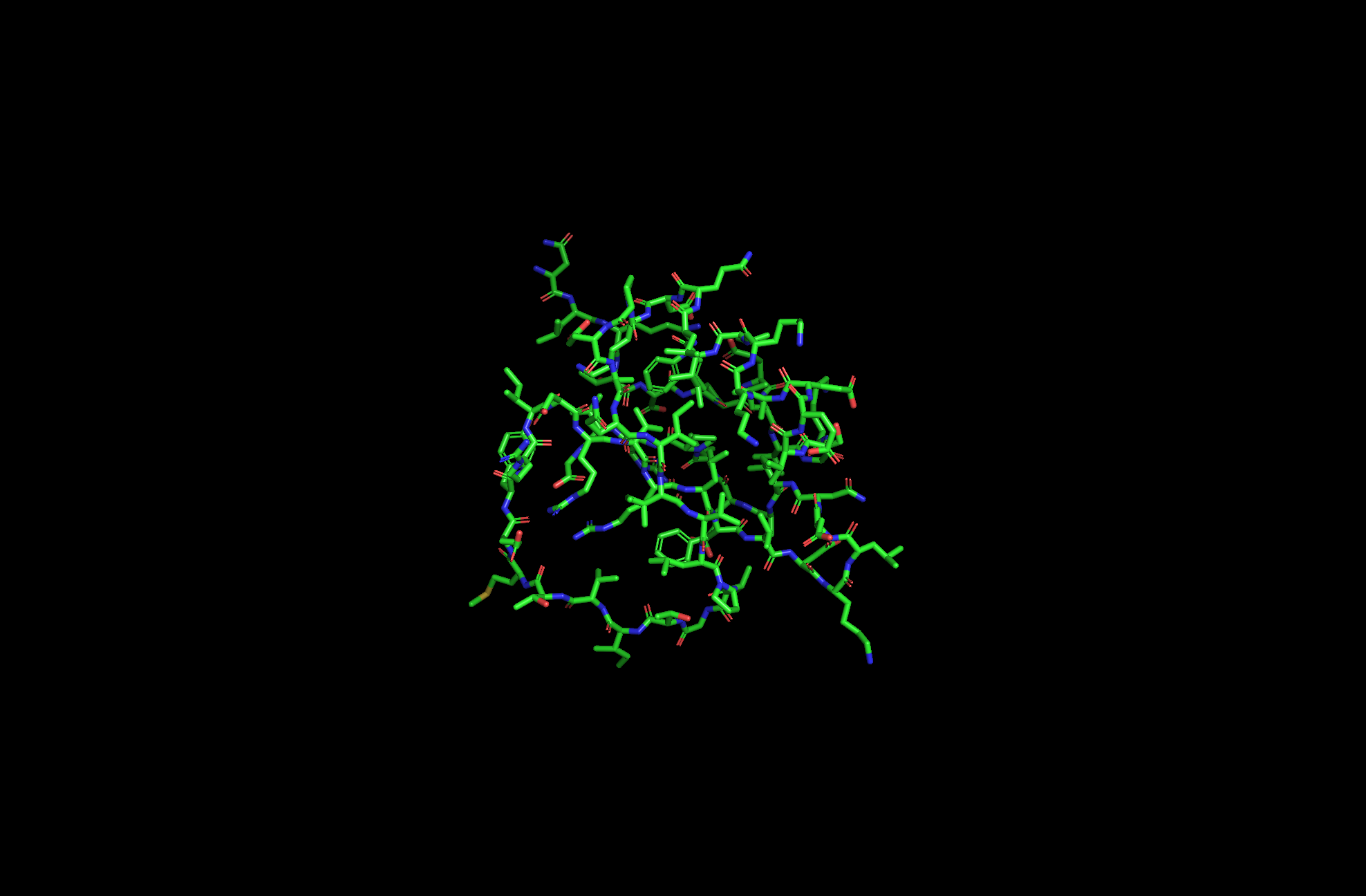
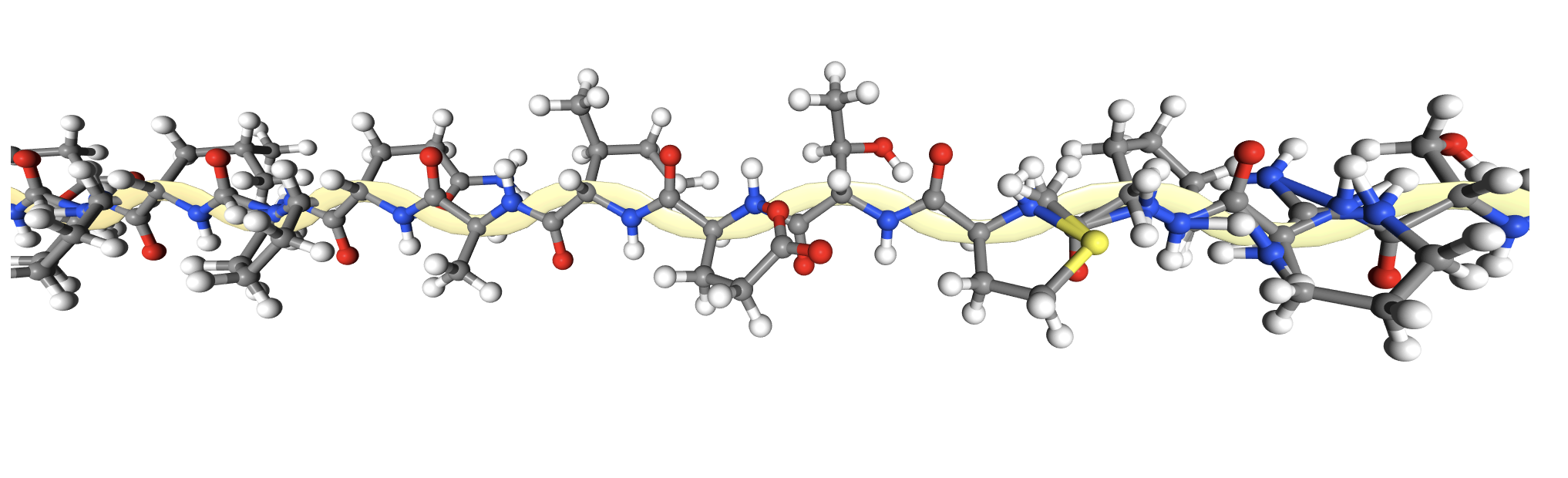
2CI2 Licorice Visualization
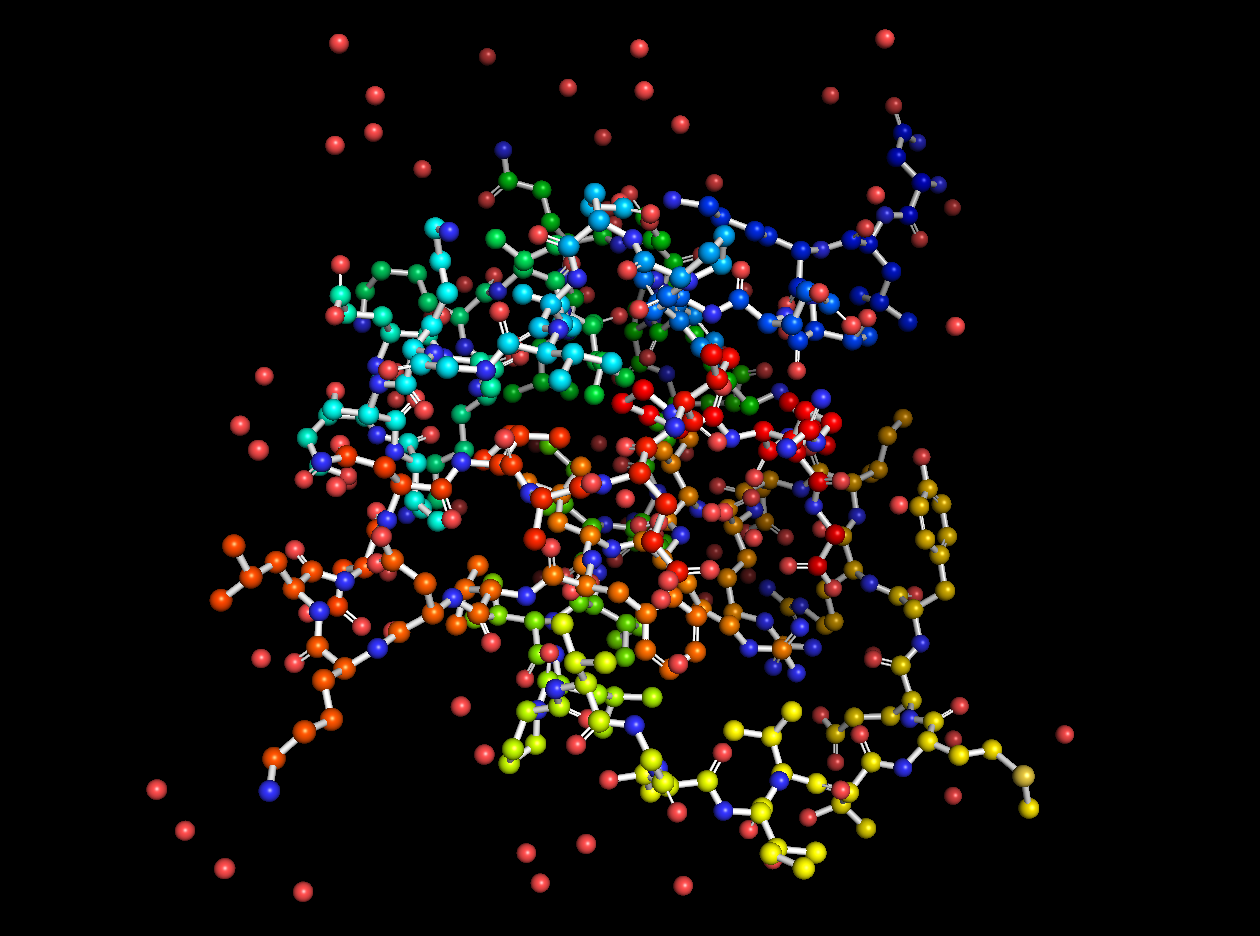
2CI2 Ball and Stick Visualization
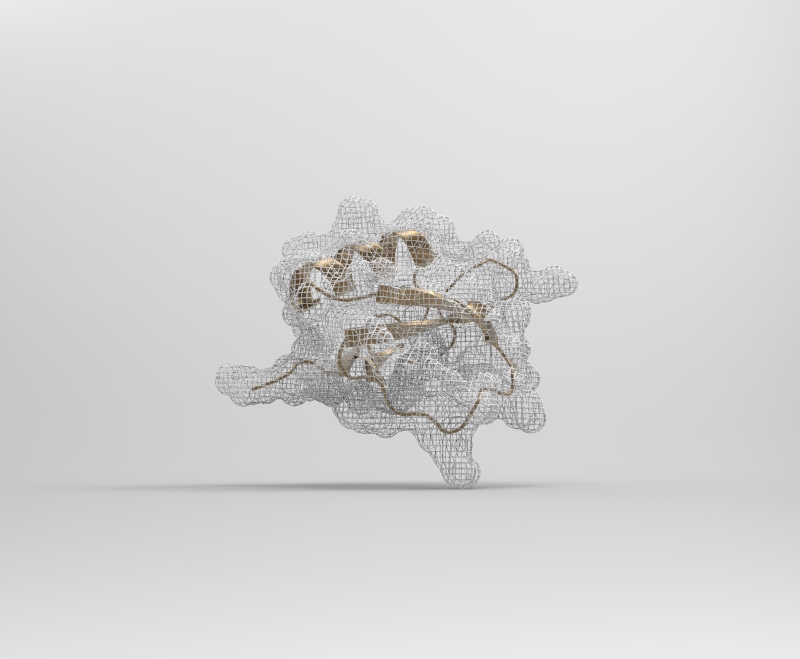
In addition to the standard modes, I exported the protein as a 3D model and rendered it for cool visuals.


Beta sheets.
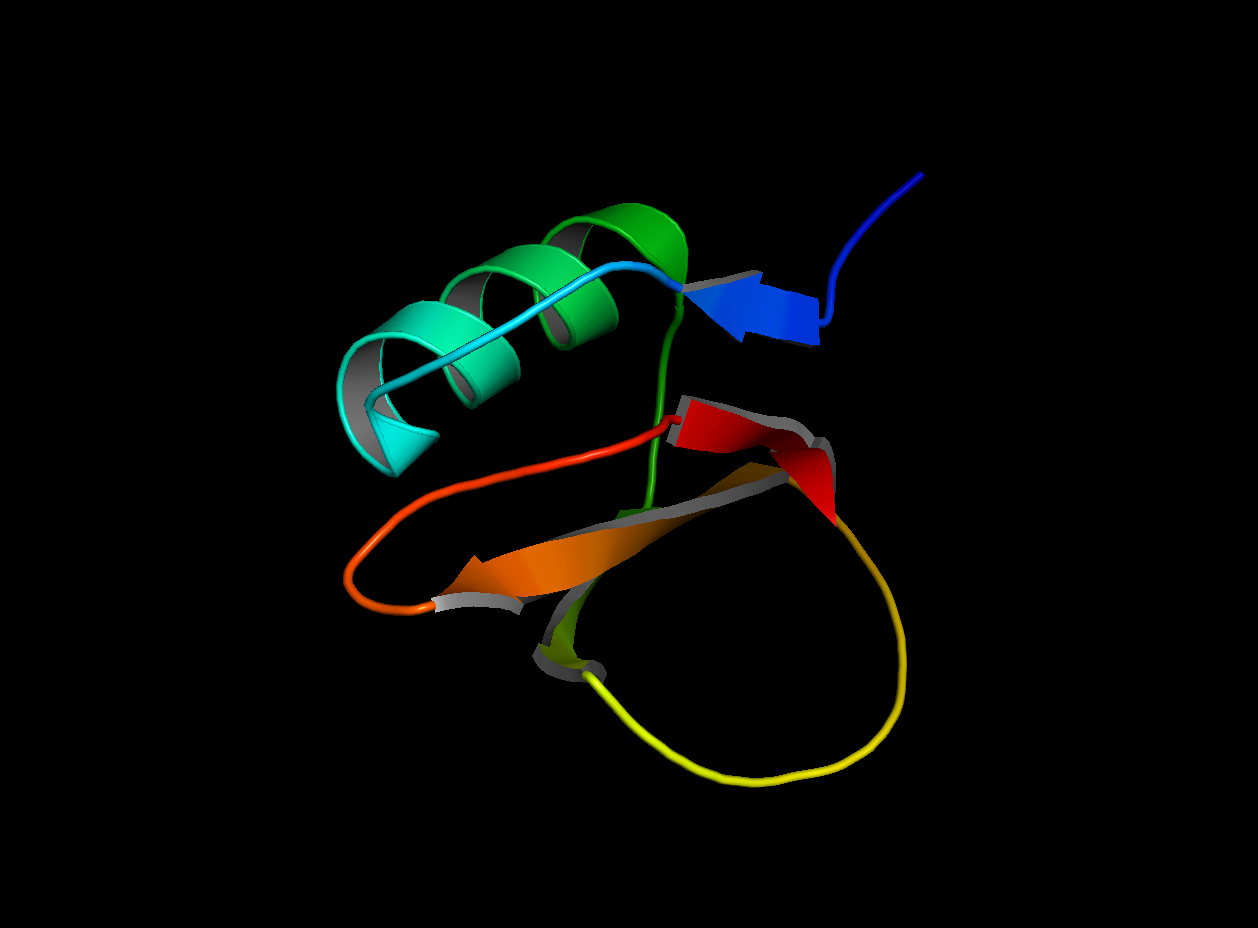

Red areas correspond to hydrophobic areas.

Yes, the protein has a hole and multiple pockets.
Part B: How to (almost) Fold (almost) Anything
- Folding a small (30 aa) peptide.
- Open the "Protein Folding with Pyrosetta" Jupyter notebook. Execute interactively the code in the notebook and answer the questions therein. When you are done, save the notebook (with the answers and all outputs) to an HTML file, and link it to your class page.
- Pick the lowest energy model and structurally (visually) compare it to the native. How close is it to the native? If its different, what parts did the computer program get wrong? Note: To compare the structures you have first to align them to the native. You can do that very easily in PyMOL. Here is a short video tutorial on aligning structures with PyMOL
- Pick the lowest RMSD model and structurally compare it to the native. How close is it to the native? If its different than the lowest energy model, how is it different? Remember that in a blind case, we will not have the benefit of an RMSD column.
- Fold your own sequence! In question 1 we used the sequence from a human protein as input to the folding algorithm. Yet, in principle, you can give any arbitrary sequence of amino acids as an input.
- Use any process to create a sequence of 30-50 amino acids, and predict it's 3D structure using the notebook from Q1. You can try to run the script with multiple parameter combinations and compare the results. Log the parameters that had the best outcome.
- Compare the resulting structures of 2(a) with those from question 1. Do the structures in both cases look protein-like ? If not, can you think of an explanation?
- Try folding multiple sequences to come up with the most protein-looking structure!
- Folding protein homologs (extra credit) For this exercise you will be running multiple protein folding simulations. If you don't have access to a powerful machine, use any of the folding servers listed in the resources.
- Take the protein sequence from question 1 and randomly change 5 letters to any other amino acid. Predict the protein structure of the unedited (probably done already in Q.1) and edited protein and compare the results. Did the changes you introduced changed the structure significantly?
- Take again the original sequence from Q.1 and now change 5 letters to favorable alternatives according to the BLOSUM matrix. Predict the protein structure for the new sequence and compare with the results of 3(a). Did the new changes have the same effect to the structure?
- By using the BLOSUM matrix as a guide, try to introduce as many changes as possible to the protein sequence, without significantly changing it's structure.
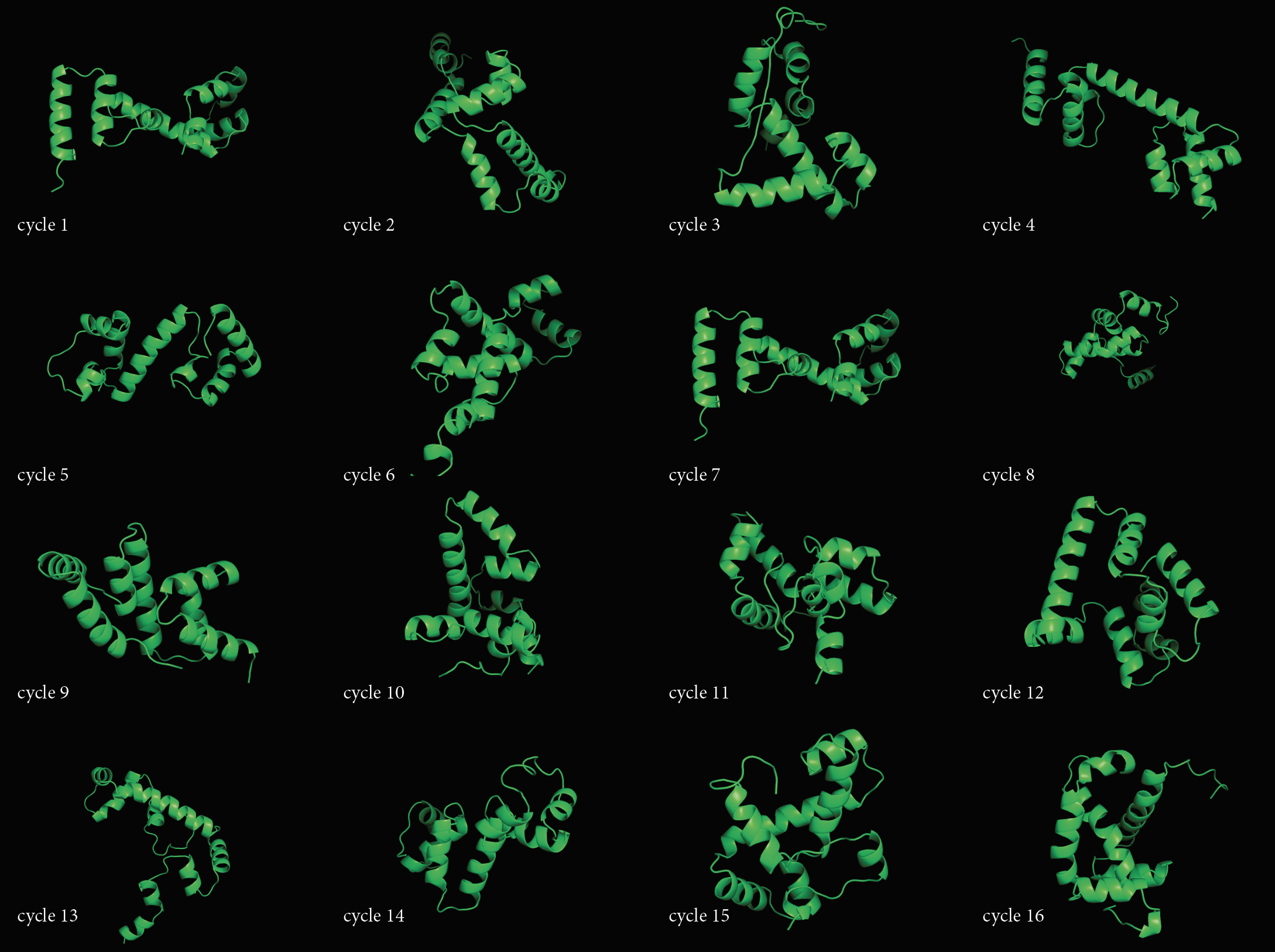
Check out the Jupyter Notebook here.