BIODESIGN
PRELAB
1. Restriction Digest
Where do PstI and EcoRV cut within their recognition sequences?
- PstI: 5'...C T G C A|G...3'
- EcoRV: 5'...G A T|A T C...3'
3'...G|A C G T C...5'
3'...C T A|T A G...5'
What gene is encoded into pPSU1 and pPSU2?
- AmpR is encoded into both plasmids.
How many copies of the plasmid would you roughly expect?
- Given the origin of replication on both plasmids is high copy number, >10.
2. DNA Libraries
How big is the E. coli genome?
- The E. coli genome is 4.6M bp.
The spectinomycin gene cassette of interest is 1.2 kb. What is the probability of finding that gene in a sheared E. coli gene fragment of 2,000 bases?
- 2000-1200/(4600000) = 0.017%
How many E. coli transformants would you have to screen through to have a 99% probability of finding the spectinomycin gene?
- (1-0.0017)^n < 0.01, so n > 26465
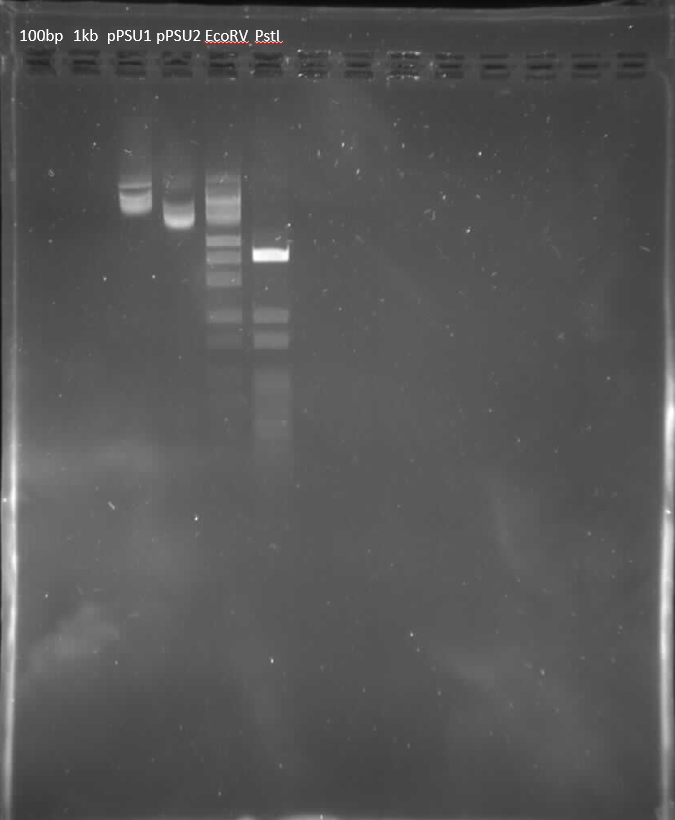
PREPARATION OF AN OPEN SOURCE DNA LADDER
- Harvested pPSU1 and pPSU2 DNA from overnight cultures using Qiagen Miniprep Kit.
- Concentration of DNA measured to be:
- pPSU1: 227.8ng/mL
- pPSU2: 334.9ng/mL
- Separate digestion reactions with NEB PstI-HF and EcoRV-HF.
- Run alongside 100bp and 1kp.

GDNA EXTRACTION WITH THE DNEASY KIT
- Harvested 3mL overnight culture of spectinomycin-resistant bacteria split into two 1.5mL tubes. gDNA extracted using DNEasy Kit from Qiagen.
- Concentration of extracted gDNA in two tubes measured to be:
- 127.1ng/mL
- 152.4ng/mL
SHEARING AND GEL ELECTROPHORESIS OF GENOMIC DNA
- Four separate restriction digests with 2ug 152.4ng/mL gDNA each:
- EcoRI-HF, EcoRV-HF, PvuII-HF
- Sau3AI stock
- 1:10 Sau3AI stock:1X CutSmart buffer
- 1:100 Sau3AI stock:1X CutSmart buffer
- Run digest at 37C for 30 minutes
- 400ug of gel were extracted using Qiagen Gel Extraction kit, yielding two 25uL tubes with 6.6ng/uL DNA.
- In an attempt to further concentrate the DNA, I used the Zymo DNA Clean & Concentrator kit on my samples. The processes reduced my yield to only 25uL of 3.6ng/uL ultra-pure DNA, likely because the DNA was not fully eluted by the elution buffer and remained in the column.

END REPAIR AND CLONING OF THE SHEARED GENOMIC DNA
- Using the maximum 19uL of sheared gDNA (for 74.1ng total), I used the NEB Quick Blunting Kit to blunt the digested DNA fragments, yielding 3.9ng/mL final blunt gDNA.
- The blunted gDNA was cloned into the linearized pMiniT 2.0 Vector using the NEB PCR Cloning Kit. Belen and I collaborated on this step to create a positive control using the control amplicon included in the kit. We also designed a negative control to be just the thawed competent cells without any plasmid.
- Ligation reaction aliquot 1
- Ligation reaction aliquot 2
- Ligation reaction aliquot 3
- Positive control
- Negative control
- The electroporation step began by transfering previously made Spec+Carb and Carb selection plates to 37C to warm. Destination eppindorfs and SOC outgrowth media were left at room temperature.
- Cuvettes, circularized plasmids, 5alpha competent cells, and eppindorf tubes for mixing were thawed on ice.
- 25mL cell aliquots were transfered to the chilled eppindorfs and mixed with 3uL of the ligation reaction (except for the negative control, which did not recieve the plasmid reaction).
- Cell plasmid mixtures were transfered to cuvettes, which were tapped gently to settle the mixture at the bottom of the well.
- For electroporation, we used 50uF capacitance, 100ohm resistance, 1.83V voltage, and aimed for a 4.2ms time constant. All my samples except for the positive control arced, yielding time constants closer to 2.44ms.
- 975uL of SOC outgrowth media were quickly mixed into the cuvette and the mixture was was transfered into a destination eppindorf.
- Destination eppindorfs were incubated on the rocker at 37C for one hour.
- Plates were colonized with 50uL or 200uL of media for each condition depending on size and the remaining electroporation mixture was transferred to liquid Spec+Carb or Carb culture.
- After 24 hours incubation rocking at 37C, plates and liquid cultures were assayed visually for growth.
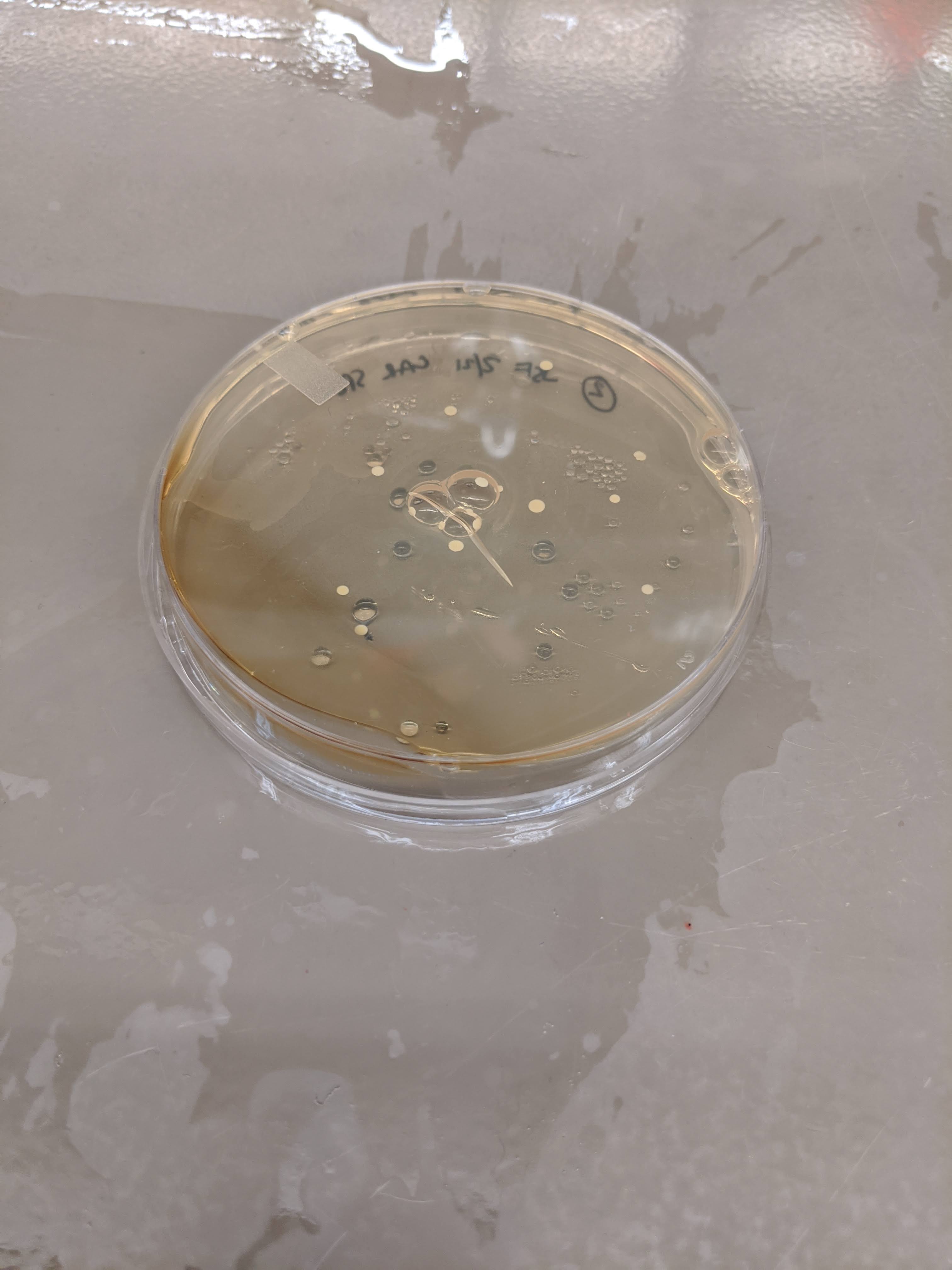
ISOLATION OF COLONIES WITH SPECTINOMYCIN RESISTANCE
- No colonies were detected on positive control plates or liquid medias for any antibiotic condition. This was surprising especially because the positive control was the only electroporation sample I ran that did not arc.
- Colonies were detected on Carb plates and Carb and Spec+Carb liquid cultures for the negative control. This was completely unexpected because our negative control was competent cells without any plasmid. We suspect either some natural resistance mechanism within 5alpha cells (which is unlikely because they are well-characterized) or human error that resulted in plasmid introduction.
- Strong colonies were detected on one Carb plate from aliquot 3, but not in any liquid culture.
- Sparse colonies were detected on two Spec+Carb plates from aliquot 2! One of these colonies will be selected for plasmid sequencing from GenWiz.
This site documents work done in HTGAA 2020.
JOE FARAGUNA