PROTEIN
PART A: PROTEIN ANALYSIS
1. General Questions
How many molecules of amino acids are in 500g of meat, given an average amino acid is 100 Daltons?
- Assuming the meat is 100% protein and given that 1Da = 1g/1 mole, there would be500g/1*1 mole/100g*6.023e23 molecules/1 mole= 3e24 amino acids.
Why are there only 20 natural amino acids?
- There are 20 natural amino acids because that number represents a balance between several factors. Although there are 43 possible codons, degeneracy within the codon to amino acid mapping scheme makes translation more robust to DNA mutations. Furthermore, tRNAs must physically recognize their designated codon reliably, and some codons are similar enough that it would be difficult for tRNAs to accurately differentiate them. Finally, by limiting the number of amino acids (and therefore the number of tRNAs) to 20, translation is less likely to stall while the ribosome waits for the correct tRNA. 20 seems to be a sufficient number for the complexity required to span a wide area in the chemical property space. For more information, see this overview article.
Why are most molecular helices right-handed?
- Most molecular helices are right-handed because chemistry and biology are chiral. Molecules with mirrored structures are not compatible with the "handedness" of the cellular environment. It is unclear how this homochirality developed to favor right over left.
Where did amino acids come from before enzymes that make them, and before life started?
- The RNA world theory suggets that life developed from complex RNA systems that performed multiple enzymatic, information-encoding, and structural functions. As these RNA molecules catalyzed their own self-replication, they may have began also to explore a mutational space that allowed them to interact with amino acids. This evolution may have eventually led to the creation of fast and precise enzymes and later complex life.
What do digital databases and nucleosomes have in common?
- Nucleosomes are a way to dynamically compress and expand DNA physically to save space or make DNA accessible for reading. Digital databases balance space efficiency and read time in a similar way. Additionally, covalent modifications to the nucleosomes themselves serve as "bookmarks" that allow the DNA to be indexed similarly to digital databases.
2. Protein selection
Protein description
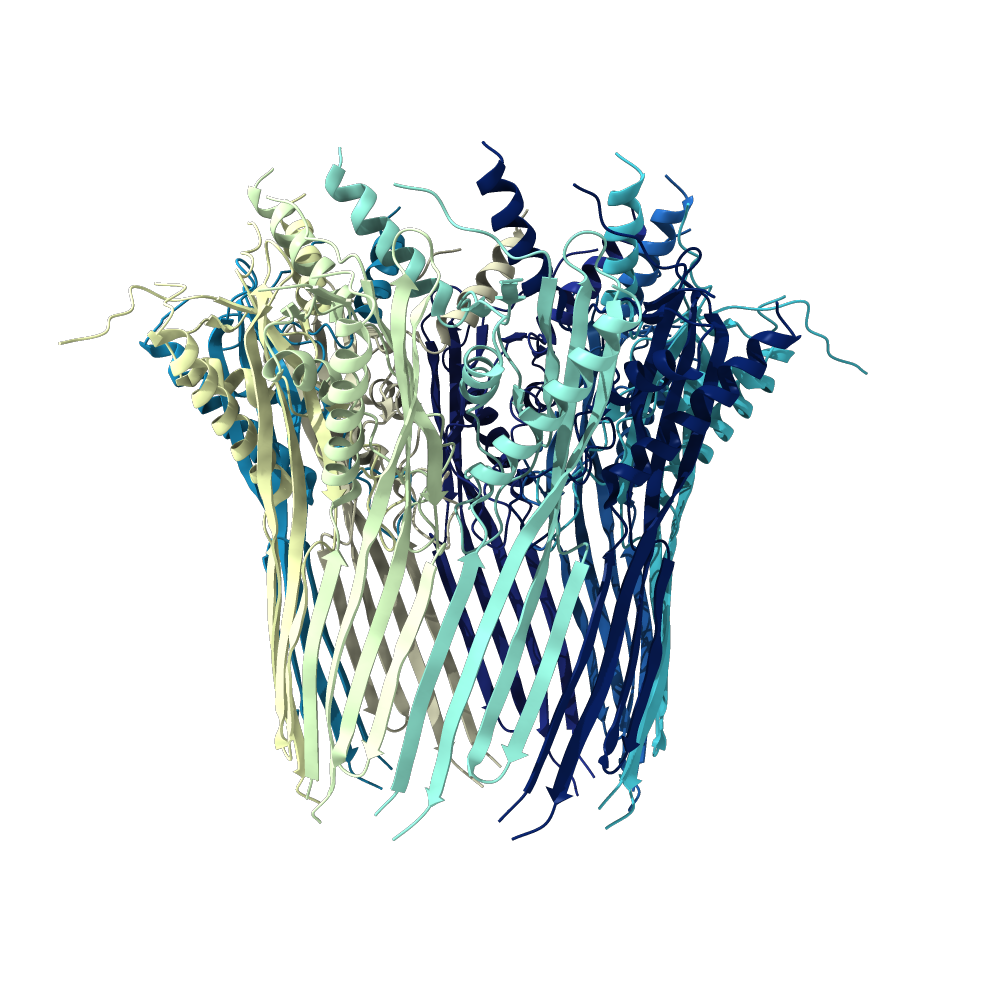
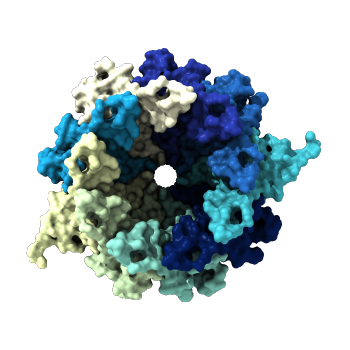
- I have selected CsgG, the outer membrane protein that secretes the two curli protein subunits A and B (PDB). The entity I've selected is in its membrane-bound conformation and is from E. coli. The CsgG protein sits extends through the outer bacterial membrane and into the periplasm. curli is an amyloid fiber produced by many forms of bacteria that is the major protein component of the extra-cellular matrix. curli fibers are strongly involved with biofilm formation, cell adhesion and are implicated in several human diseases (paper). The outer membrane protein is not involved in these functions, but it's structure is more intricate than the A and B subunits, so I selected it for further investigation.
How long is it? What is the most frequent amino acid?
- CsgG is 2538aa long with 9 identical side chains (D, A, B, C, E, F, G, H, I) all of 277aa in length. The most common amino acid in the whole protein is Leucine (261). Each side chain has 29 Leucines.
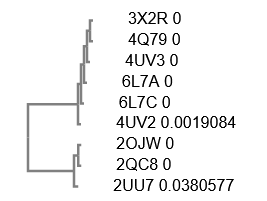
How many protein sequence homologs are there?
- I identified 9 protein sequence homologs to 3X2R, the CsgG protein of interest.
Does your protein belong to any protein family?
- CsgG is an outer membrane secretion channel protein.
When was the structure solved? Is it a good quality structure?
- The structure was deposited in 2014 and was solved using X-ray diffraction at a resolution of 2.9A. wwPDB Validation ranks in the low percentile compared to X-ray structures of a similar resolution for most of its metrics.
Are there any other molecules in the solved structure apart from the protein?
- There are no other molecules in the solved structure apart from the protein. However, CsgG often appears in a complex with CsgF, an intracellular protein that allows for efficient CsgA transport. The two proteins together bind their substrate the CsgAN6 peptide, so other solved structures include these molecules.
Does your protein belong to any structure classification family?
- CsgG is part of the TolB N-terminal domain-like superfamily (SCOP).
3. Protein visualization
CsgG has a similar number of α-helices and Β-sheets distributed in different locations. Technically CsgG contains a 36 stranded Β-barrel. The Β portion of the protein sits within the plasma membrane, while the bulkier α portion interacts with proteins within the periplasm (Frontiers). The exterior surface of the protein is quite hydrophobic, which makes sense as it is membrane-bound. There is a clear channel that runs throughout the center of the protein, allowing for transport across the outer membrane.
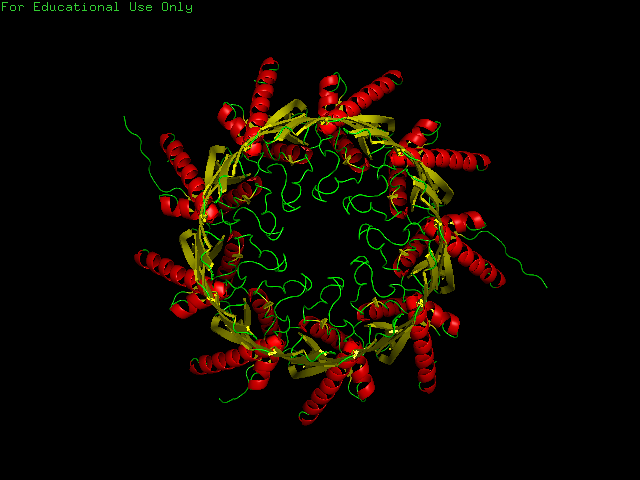
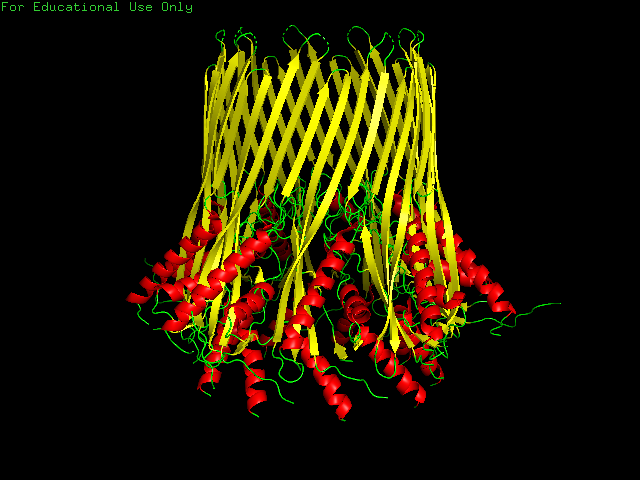


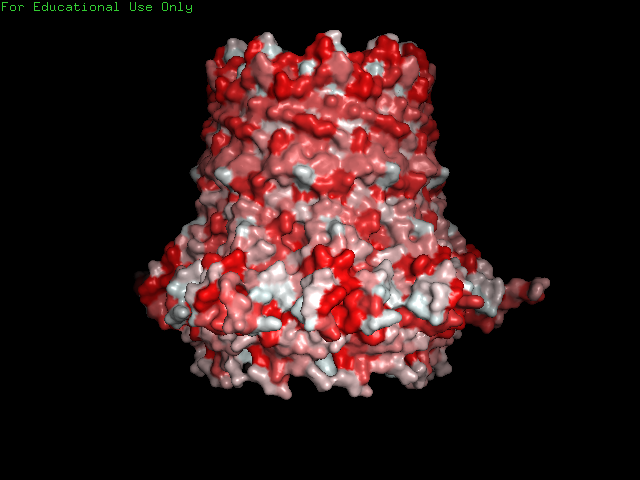
PART B: HOW TO FOLD (ALMOST) ANYTHING
1. Folding a small (30aa) peptide
Visualize the centroid-only structure and see the difference with the full atom that we visualized above? Print again the information for the first residue and compare?
- Both the centroid-only structure and the full atom share the same linear structure. The full atom includes the individual atoms themselves for improved resolution.
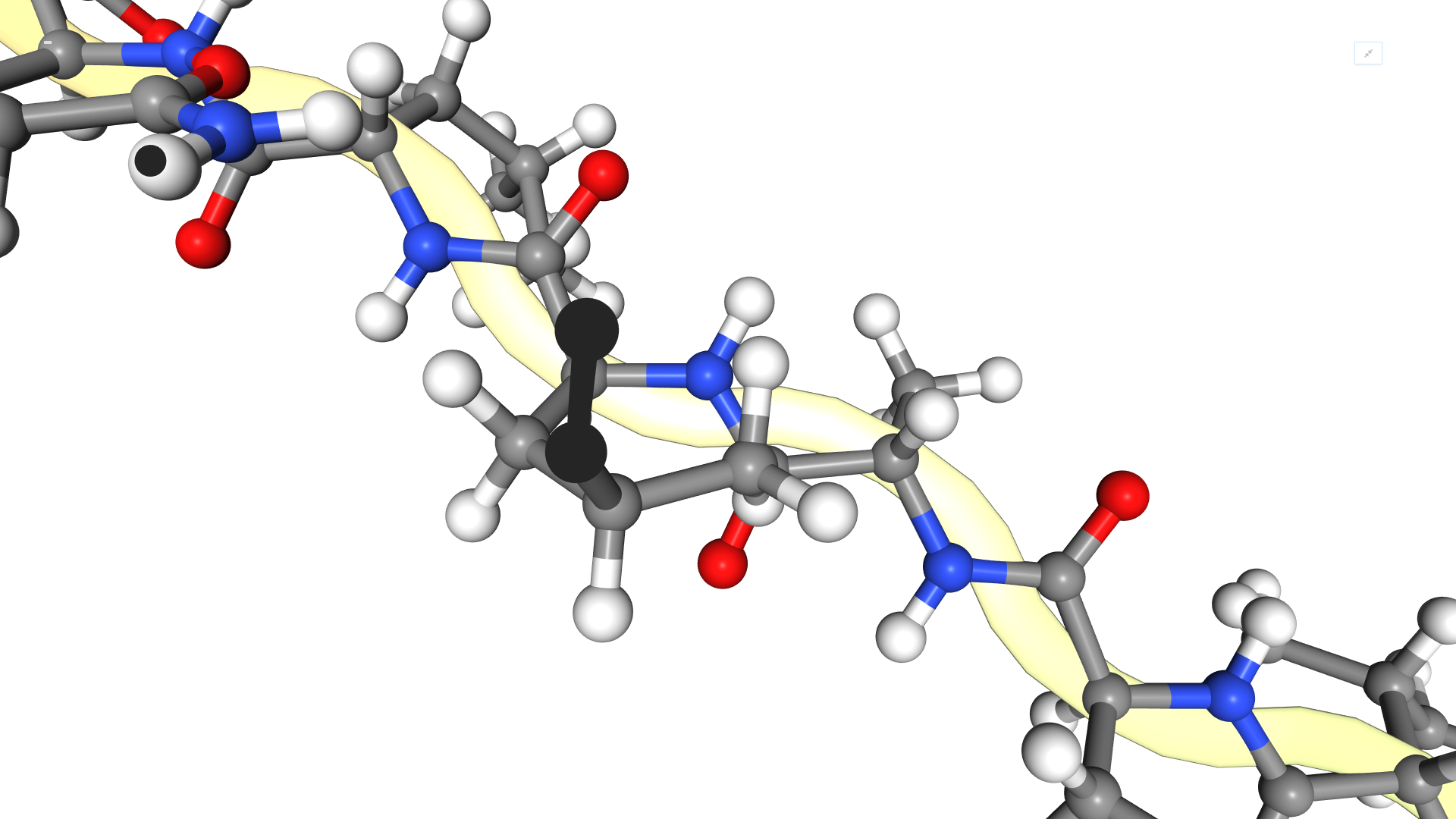
-
Centroid Atom Residue 1: ASP:NtermProteinFull (ASP, D):
Base: ASP
Properties: POLYMER PROTEIN CANONICAL_AA LOWER_TERMINUS POLAR CHARGED NEGATIVE_CHARGE ALPHA_AA L_AA
Variant types: LOWER_TERMINUS_VARIANT
Main-chain atoms: N CA C
Backbone atoms: N CA C O H
Side-chain atoms: CB CENResidue 1: ASP:NtermProteinFull (ASP, D):
Base: ASP
Properties: POLYMER PROTEIN CANONICAL_AA LOWER_TERMINUS SC_ORBITALS POLAR CHARGED NEGATIVE_CHARGE METALBINDING ALPHA_AA L_AA
Variant types: LOWER_TERMINUS_VARIANT
Main-chain atoms: N CA C
Backbone atoms: N CA C O 1H 2H 3H HA
Side-chain atoms: CB CG OD1 OD2 1HB 2HB
Atom Coordinates:
N : 0, 0, 0
CA : 1.458, 0, 0
C : 2.00885, 1.42017, 0
O : 1.25096, 2.39022, -2.58987e-16
CB : 1.99452, -0.771871, -1.208
CEN: 2.35051, -1.69379, -1.45468
H : -0.5, -0.433013, -0.75
Mirrored relative to coordinates in ResidueType: FALSEAtom Coordinates:
N : 0.229, 36.012, 74.172
CA : 0.041, 35.606, 75.594
C : -0.096, 36.849, 76.498
O : -0.951, 36.895, 77.382
CB : 1.225, 34.718, 76.092
CG : 2.159, 34.156, 74.999
OD1: 1.688, 33.361, 74.151
OD2: 3.378, 34.497, 75.007
1H : 1.056, 35.74, 73.68
2H : -0.43, 35.723, 73.478
3H : 0.251, 36.981, 73.928
HA : -0.884, 35.037, 75.696
1HB : 1.839, 35.199, 76.854
2HB : 0.67, 33.892, 76.539
Mirrored relative to coordinates in ResidueType: FALSE
How many 9-mer and 3-mer fragmets do we have in our database?
- 200 9mer and 200 3mer fragments.
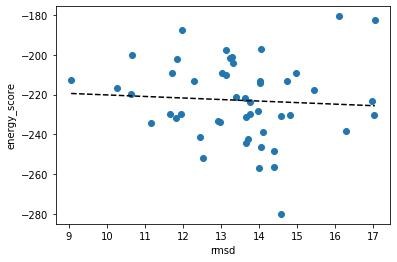
Plot the energy_score VS rmsd. Are the lowest energy structures the closest to the native one?
- The lowest energy structures are not always closest to the native one. RMSD and energy were almost completely uncorrelated, with a Pearson correlation coefficient of -0.065532.
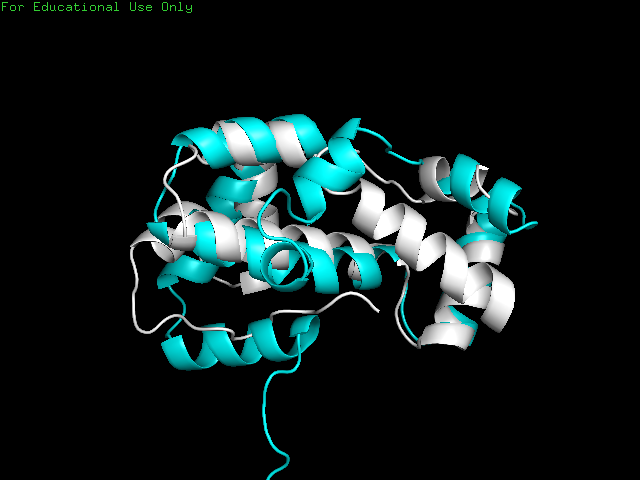
Although the native and closest predicted folded proteins were similar in structure, they were not identical.
2. Fold your own sequence!
Sequence generation
- I encoded my name into amino acids as LLQE FARAGVNA and repeated it several times to generate a 116aa protein:
LLQEFARAGVNALLQEFARAGVNALLQEFARAGVNALLQEFARAGVNALLQEFARAGVNALLQEFARAGVNALLQEFARAGVNALLQEFARAGVNALLQEFARAGVNALLQELLQE - I ran the notebook with default parameters and with the kmer libraries Thras generated for us for the known protein folding. The predicted foldings had energy scores similar to the previous protein, ranging from -250 to -170. I selected the protein with the lowest energy score as the best folding. As expected, name motifs are repeated
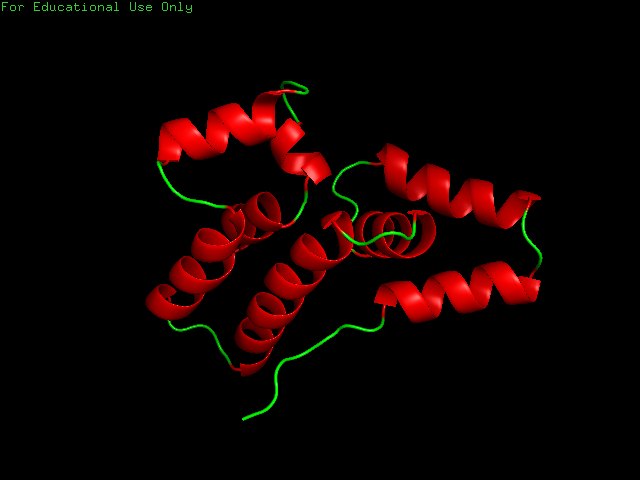
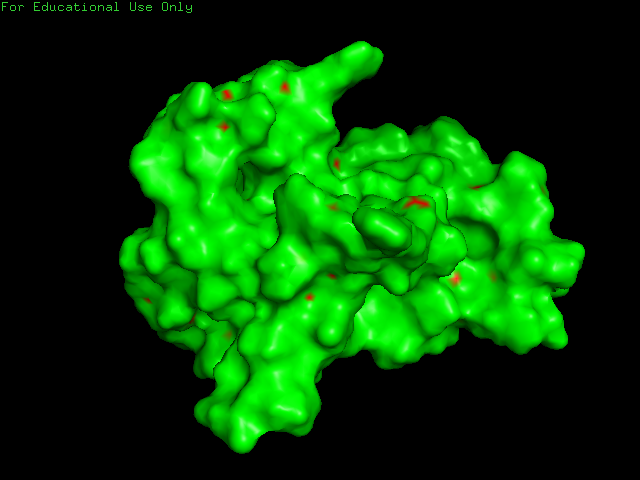
Compare the resulting structures of 2(a) with those from question 1. Do the structures in both cases look protein-like ? If not, can you think of an explanation?
- Both structures appear protein-like with repeated motifs of α-helices and loops.
This site documents work done in HTGAA 2020.
JOE FARAGUNA