Week 2: Bio Design, Making a DNA Ladder
DNA ladders are a measurement tool for DNA. By using electrophoresis, DNA is pulled through an agarose gel from negative to positive. Different parts of the DNA are caught by the gel's traction due to their particular molecular weights, and so create a "ladder" effect in the gel. The measures on a DNA ladder are made by using specific DNA which are known to stop at particular intervals, eg. 100 basepairs, 200bp, 400bp, etc. This week we used a DNA ladder method from the Tan Lab at Penn State University, which utilizes two plasmids, pPSU1 and pPSU2 with two common restriction enzymes, EvoRV and PstI (which cut the DNA into ordered fragments), to create a DNA ladder which is much cheaper to produce than commercial offerings.
In my understanding so far, plasmids are virus-like molecules which is basically a little ring of DNA. A fragment of DNA of interest can be inserted into the plasmid, and because a plasmid will insert itself into a bacterium like e. coli, the multiplying bacteria effectively becomes a cloning factory for the plasmid with the inserted DNA. In our case, the DNA insert was amphicilin antibiotic resistance, into two plasmids, pPSU1 and pPSU2.
In silico (Using Addgene)
Distance between PstI sites in pPSU1:
- 1696 (500) 2196 (1000) 4196 (1000) 5196 (700) 5896 (800) 6696 (900) 7596
Distance between EcoRV sites in pPSU1:
- 241 (500) 741 (1000) 1741 (1500) 3241 (2000) 5241
Distance between PstI sites in pPSU2:
- 1500 50 100 200 300 400 500 600
Distance between EcoRV sites in pPSU2:
- 750 3000 4000
The plasmids are encoded with AmpR, which confirms amphicilin resistance, giving the e. coli greater fitness to multiply over other bacteria.
ColE1/pMB1/pBR322/pUC are different origins of replication (ORI). According to open wetware pUC has a copy number of 5-700, while pBR322 only has a copy number of 14-20. Addgene's spec for pPSU1/2 says pUC9, and "High copy", which would suggest somewhere in the hundreds.
How do the plasmids' replication relate to that of pBR322 (genbank)?
pBR322 was taken from replication elements of pMB1, and is closely related to pUC, being highly similar except for one mutation and the absence of a rop gene, which gives it its high copy number. The rop gene encodes a restrictor on plasmid copy number. Both pBR322 and pUC19 are derived from the origin of replication of pMB1.
Lab work
We begin by taking Penn State's plasmids, pPSU1 and pPSU2, which have already been incubated with e. coli, and initiate the process of isolating the DNA. Using the ZymoPure plasmid miniprep kit, we lyse the e.coli, breaking down its membrane, before neutralizing the lysate. A bit like cooking. Then we added various buffers, like a binding buffer which binds the DNA with the ZymoSpin II Column (a kind of tiny sieve for getting rid of all the other gunk that isn't DNA). After a lot of washing, and getting rid of residue, we have a column with ostensibly pure DNA which is diluted into 25ul of water.
These two samples of 25ul DNA were measured on the nano spectrophotometer, which determined the concentration of DNA by its absorbance of a certain wavelength.
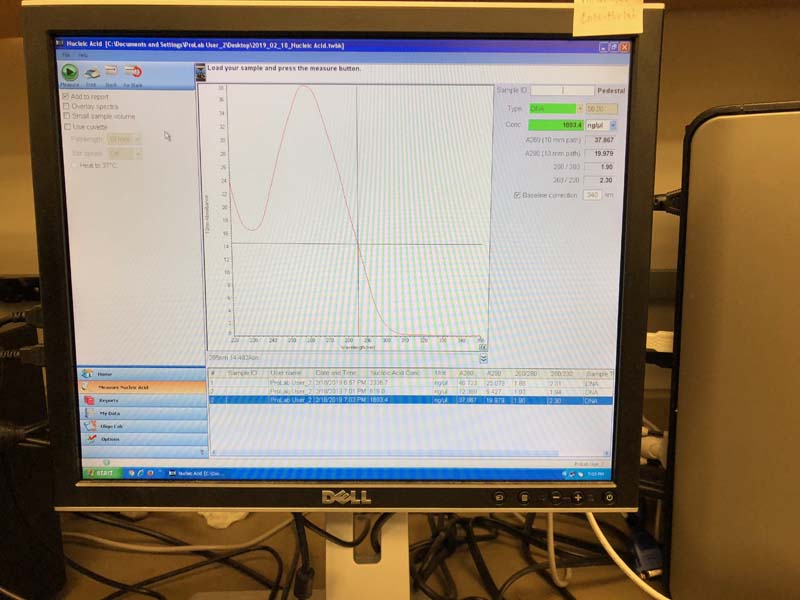
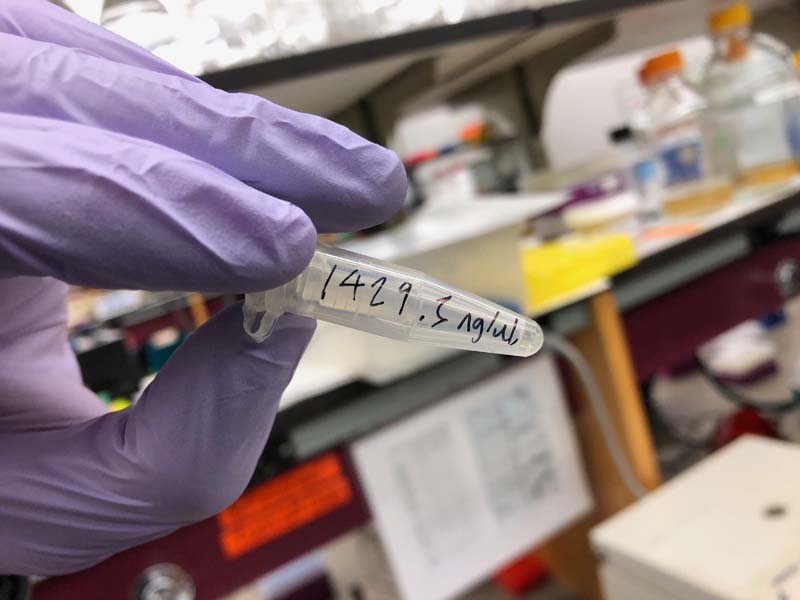
Mine came out at concentrations of 1863 and 1429.3ng/ul. Everyone's concentrations were quite high, apparently.
In the second session, we took the DNA and prepared it in a dilution with two restriction enzymes, EcoRV and PstI, which are the enzymes that "cut" the string of DNA at particular points, calibrated to create a ladder at particular lengths. We also prepared four controls, of the two DNA samples, but uncut, and of commercial reference ladders.

Had a bit of confusion around the dilution process. In order to dilute teh 1429.3ng/ul pPSU2 to 100ng/ul in a 50ul solution, we first diluted the DNA at 2 parts DNA to 18 parts water, in order to make a 20ul dilution with a concentration of 142.9ng/ul (10x), or 0.1429ug/ul. In order to get the 1ug of DNA into the 50ug reaction... 1/0.143 = 7.04ug. Then 50ug - 7 to account for the total volume of the reaction.

Then we incubate them all for 15min at 37C, and added 10ug of a loading dye to the mixture. We also prepared the controls.

The incubator.

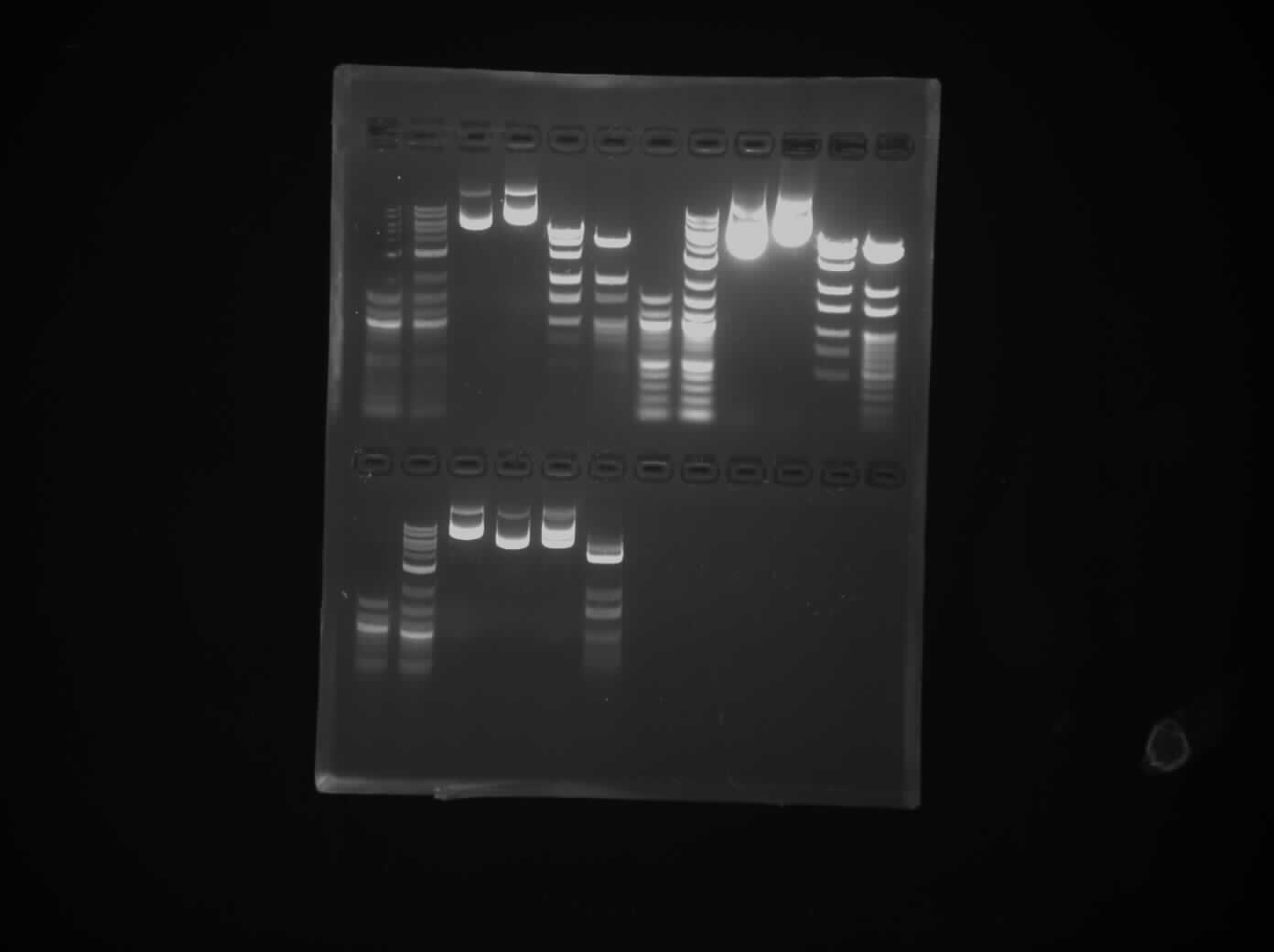
These solutions were pipetted into slots in the agarose gel, which is covered in a layer of water. The power supply is connected, with the DNA lanes travelling from the cathode to the anode (negative to postive). The gel box is run at 120V (140V for accelerated movement) for around an hour. You can see bubbles showing that it's working.


Finally the agarose slab is taken for UV imaging. We had a fairly successful run, I messed up one of my reactions (the PstI) but you can see from the images that they are pretty identical to the commercial ladders.